23+ 顶刊纯公开数据库建模!病理切片预测基因表达与患者治疗反应,原文提供了非常清晰的代码实现!
通过对16个癌症类型的大规模数据集进行验证,ENLIGHT–DeepPT在多种独立患者队列中成功预测了真实的免疫治疗反应者。
图像预测基因表达
病理学长期以来被认为是癌症临床诊断和预后的金标准。随着数字病理学的快速进步,机器学习和人工智能方法在图像分析中的应用,使得从肿瘤切片中提取临床相关信息成为可能。利用这些技术,全视野切片图像(WSI)结合苏木精-伊红(H&E)染色的组织切片已经用于计算机化的各类研究,这包括:肿瘤诊断、癌症类型分类、区分高低突变负荷的肿瘤、识别基因突变、预测患者生存、检测DNA甲基化模式和有丝分裂、以及量化肿瘤免疫浸润和空间免疫细胞浸润等等。

▲ TCGA数据库中肿瘤样本的病理染色切片
2024年7月3日,来自美国癌症研究所(NCI)的研究人员在Nature Cancer[23.5]期刊上提出了一种名为ENLIGHT–DeepPT的深度学习框架,旨在通过组织切片图像预测癌症治疗的反应。文章中,研究团队详细介绍了如何利用DeepPT模型预测肿瘤基因表达,并结合ENLIGHT模型预测患者对靶向和免疫治疗的反应。通过对16个癌症类型的大规模数据集进行验证,ENLIGHT–DeepPT在多种独立患者队列中成功预测了真实的免疫治疗反应者。

▲ 10.1038/s43018-024-00793-2
小编先前冲浪的时候看到了这篇文章,其网络结构与设计并不复杂,并且没有任何的实验内容。整体讲讲,DeepPT 包括了四个主要组成部分:
-
(i) 图像预处理:将每张全幅切片图像分割成小块/补丁,仅选择包含组织的块,排除背景区域。包括颜色标准化,以最小化染色变化(异质性和批次效应)。
-
(ii) 特征提取:使用预训练的 ResNet50 CNN 模型从图像块中提取特征。通过这一过程,每个图像块由 2,048 个衍生特征(预训练 ResNet 特征)表示。
-
(iii) 特征压缩:通过自编码器网络将2,048个预训练ResNet特征压缩为512个特征。
-
(iv) 预测表达:一个两层神经网络该组件以自编码器特征作为输入,以基因表达作为输出。
-
(v) 预测患者治疗反应:使用ENLIGHT方法预测患者对靶向和免疫治疗的反应

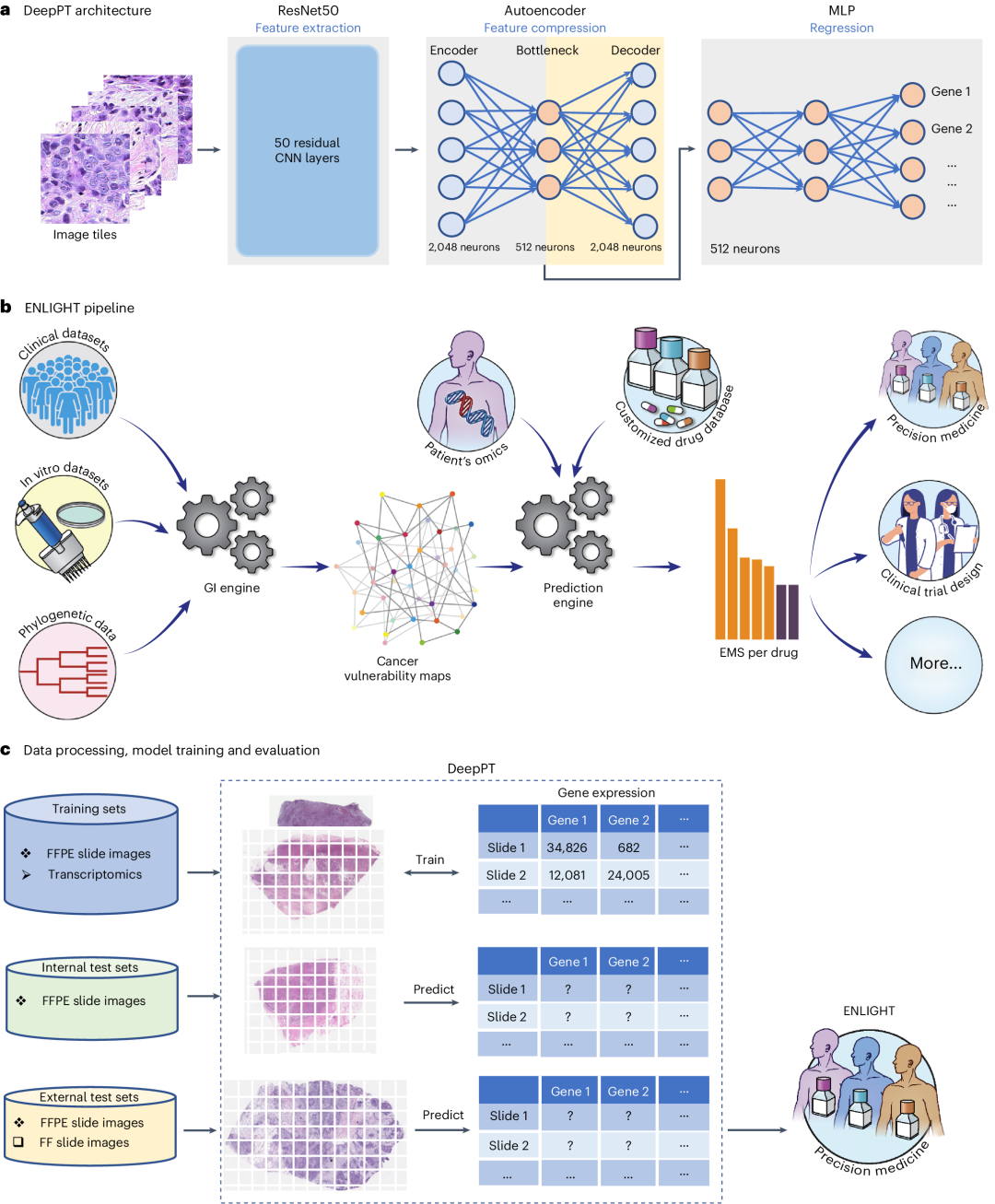
▲ a. DeepPT 架构的三个主要组件:预训练的 ResNet50 卷积神经网络(CNN)单元(左)从切片图像中提取组织病理特征;自编码器(中)将 2,048 个特征压缩到 512 个特征的较低维度;MLP(右)整合这些组织病理特征以预测样本的基因表达。b. ENLIGHT 流程概述(插图取自文献47):ENLIGHT 从各种癌症体外和临床数据源中推断给定药物的 GI 伴侣。基于 SL 和 SR 伴侣以及给定患者样本的转录组数据,ENLIGHT 计算药物匹配评分,用于预测患者的药物响应。在这里,ENLIGHT 使用 DeepPT 预测的表达数据为每个研究的患者生成药物匹配评分。c. 使用 DeepPT 和 ENLIGHT 的分析概述。顶部行,DeepPT 使用 FFPE 切片图像和 TCGA 中不同癌症类型的匹配转录组数据进行训练。中间行,训练阶段后,将模型应用于内部(保留的)TCGA 数据集和两个未进行训练的外部数据集,以预测基因表达。底部行,预测的肿瘤转录组数据在五个独立测试临床数据集中作为输入,供 ENLIGHT 预测患者的治疗响应并评估整体预测准确性。
上面所说的ENLIGHT是另一篇文章开发的,这里我们不细讲。主要还是来看看作者是怎么通过图像预测基因表达的:
① 图像预处理:openslide 、utils_color_norm以及 PIL 库分别用来图像读取以及处理显微镜切片。
② 基于最常用的torchvision库,用预训练好的ResNet50网络进行特征提取(代码看着并不复杂,只是简单的加载他人预训练的模型ResNet50_IMAGENET1K_V2.pt):
model = Feature_Extraction(model_type="load_from_saved_file")
model.to(device)
model.load_state_dict(torch.load("../ResNet50_IMAGENET1K_V2.pt", map_location=device))
model.eval()
batch_size = 64
torch_resize = transforms.Resize(224)
tiles_list_resized = []
for i in range(n_tiles):
tiles_list_resized.append(torch_resize(tiles_list[i]))
tiles_list = tiles_list_resized
data_transform = transforms.Compose([transforms.ToTensor(),
transforms.Normalize(mean=[0.485, 0.456, 0.406], std=[0.229, 0.224, 0.225])
])
def extract_features_from_tiles(tiles_list):
tiles = [data_transform(tile).unsqueeze(0) for tile in tiles_list]
tiles = torch.cat(tiles, dim=0)
features = []
for idx_start in range(0, len(tiles), batch_size):
idx_end = idx_start + min(batch_size, len(tiles) - idx_start)
feature = model(tiles[idx_start:idx_end])
features.append(feature.detach().cpu().numpy())
return np.concatenate(features)
if extract_pretrained_features:
features = extract_features_from_tiles(tiles_list)
np.save(f"{path2features}{slide_name}.npy", features)
这个预训练的ResNet50模型是使用ImageNet数据库进行训练的,后者是一个大型视觉数据库,包含了约1400万张自然图像,用于各类文章训练深度学习模型进行图像分类。通过使用在ImageNet上训练好的ResNet50模型,作者从图像切片中提取了2048个特征。
③ 使用自编码器降维特征,从2048到512,网络的结构如下:
class AutoEncoder(nn.Module):
def __init__(self, n_inputs, n_hiddens, n_outputs):
super(AutoEncoder, self).__init__()
self.encoder = nn.Sequential(
nn.Linear(n_inputs,n_hiddens),
nn.ReLU()
)
self.decoder = nn.Sequential(
nn.Linear(n_hiddens,n_outputs),
nn.ReLU()
)
def forward(self, x):
x = self.encoder(x)
x = self.decoder(x)
return x
竟然是个一层的自编码器,泪目了
④ 使用上一步自编码器提取的512个特征,预测基因表达。模型使用的是只有两层的神经网络,看代码注释我相信作者也曾经努力试过1维的卷积:
class MLP_regression(nn.Module):
def __init__(self, n_inputs, n_hiddens, n_outputs, dropout, bias_init):
super(MLP_regression, self).__init__()
self.layer0 = nn.Sequential(
#nn.Conv1d(in_channels=n_inputs, out_channels=n_hiddens,kernel_size=1, stride=1, bias=True),
nn.Linear(n_inputs,n_hiddens),
#nn.ReLU(), ## 2020.03.26: for positive gene expression
nn.Dropout(dropout)
)
#self.layer1 = nn.Conv1d(in_channels=n_hiddens, out_channels=n_outputs,kernel_size=1, stride=1, bias=True)
self.layer1 = nn.Linear(n_hiddens, n_outputs)
## ---- set bias of the last layer ----
if bias_init is not None:
self.layer1.bias = bias_init
##-------------------------------------
def forward(self, x):
#print("x.shape - input of forward:", x.shape) ## [n_tiles,512]
x = self.layer0(x)
x = self.layer1(x)
#print("x.shape - before mean:", x.shape) ## [n_tiles,512]
x = torch.mean(x, dim=0) ## sum over tiles
#print("x.shape -- after mean:", x.shape) ## [n_genes]
return x ## predicted gene_values [n_genes]
-
损失函数很简单,一个自编码器的重构损失,一个MLP的
MSELoss。
整体并不难实现
如果通过图像把基因表达预测出来了
后续想再接什么其它任务
那就很灵活了
发点小文章绝对够顶了
感兴趣的铁子们可以去复现看看
源码在正文提供了链接

开放原子开发者工作坊旨在鼓励更多人参与开源活动,与志同道合的开发者们相互交流开发经验、分享开发心得、获取前沿技术趋势。工作坊有多种形式的开发者活动,如meetup、训练营等,主打技术交流,干货满满,真诚地邀请各位开发者共同参与!
更多推荐
 19
19 0
0- 0



 已为社区贡献4条内容
已为社区贡献4条内容

所有评论(0)