MOVICS:分子分型一站式R包(01)
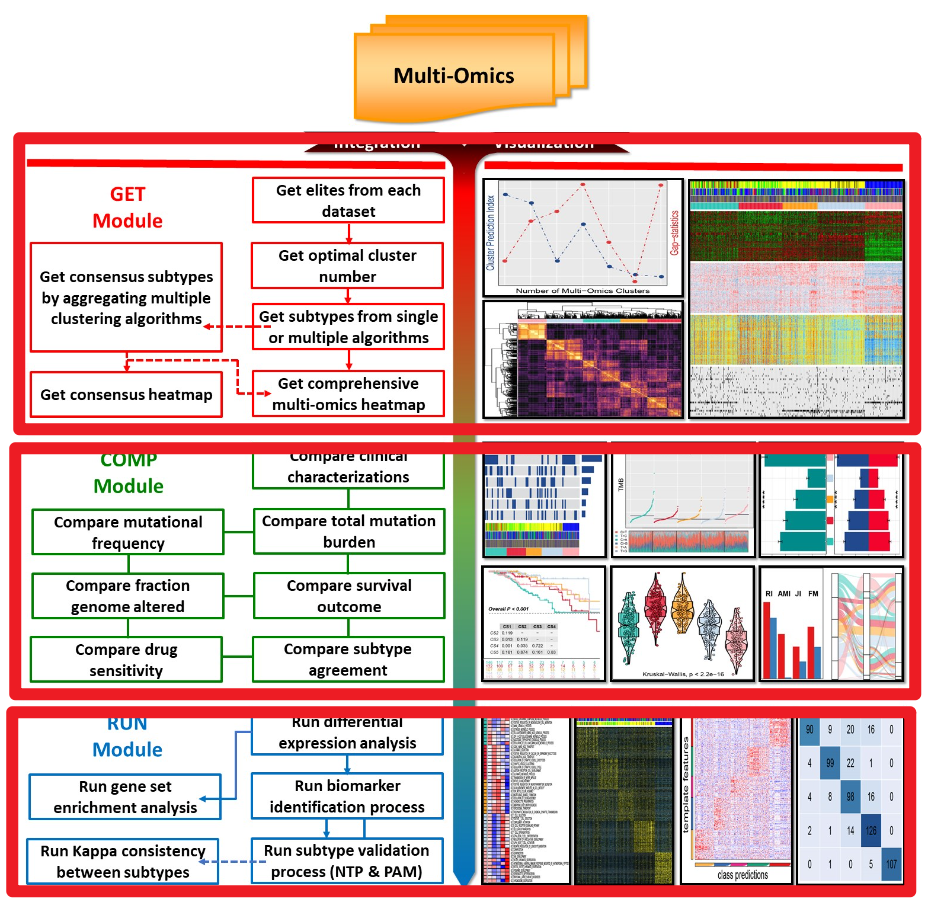
免疫浸润结果分子分型MOVICS。该包与其他分子分型R包最大的不同是它能同时使用多组学的数据,普通的分子分型R包只能通过一种组学数据进行分析,比如只能通过mRNA的表达矩阵进行分析。但是这R包它可以同时通过比如说mRNA、lncRNA、甲基化数据、突变数据进行分型。之外,它还提供了分型之后每个亚型的探索以及每个亚型内的分析。所以说这是一个一站式的包。这个的功能主要分为三个部分,示意图如下:第一个部
简介
分子分型一直是生信数据挖掘的热门技能,用于分子分型的算法非常多,比如大家常见的非负矩阵分解、一致性聚类、PCA等,一致性聚类我们在之前也介绍过了:免疫浸润结果分子分型
今天给大家介绍一个一站式的分子分型R包:MOVICS。
该包与其他分子分型R包最大的不同是它能同时使用多组学的数据,普通的分子分型R包只能通过一种组学数据进行分析,比如只能通过mRNA的表达矩阵进行分析。但是这R包它可以同时通过比如说mRNA、lncRNA、甲基化数据、突变数据进行分型。
之外,它还提供了分型之后每个亚型的探索以及每个亚型内的分析。所以说这是一个一站式的包。这个的功能主要分为三个部分,示意图如下:

第一个部分是根据不同的组学数据进行分型。大部分是比较不同的分型。第三个部分是对每个分型进行探索,以及获得每个分型特异性的分子。
每个部分包含的主要函数如下,下面会介绍:
-
GET Module: get subtypes through multi-omics integrative clustering
- getElites(): get elites which are those features that pass the filtering procedure and are used for analyses
- getClustNum(): get optimal cluster number by calculating clustering prediction index (CPI) and Gap-statistics
- getalgorithm_name(): get results from one specific multi-omics integrative clustering algorithm with detailed parameters
- getMOIC(): get a list of results from multiple multi-omics integrative clustering algorithm with parameters by default
- getConsensusMOIC(): get a consensus matrix that indicates the clustering robustness across different clustering algorithms and generate a consensus heatmap
- getSilhouette(): get quantification of sample similarity using silhoutte score approach
- getStdiz(): get a standardized data for generating comprehensive multi-omics heatmap
- getMoHeatmap(): get a comprehensive multi-omics heatmap based on clustering results
-
COMP Module: compare subtypes from multiple perspectives
- compSurv(): compare survival outcome and generate a Kalan-Meier curve with pairwise comparison if possible
- compClinvar(): compare and summarize clinical features among different identified subtypes
- compMut(): compare mutational frequency and generate an OncoPrint with significant mutations
- compTMB(): compare total mutation burden among subtypes and generate distribution of Transitions and Transversions
- compFGA(): compare fraction genome altered among subtypes and generate a barplot for distribution comparison
- compDrugsen(): compare estimated half maximal inhibitory concentration (IC50
) for drug sensitivity and generate a boxviolin for distribution comparison - compAgree(): compare agreement of current subtypes with other pre-existed classifications and generate an alluvial diagram and an agreement barplot
-
RUN Module: run marker identification and verify subtypes
- runDEA(): run differential expression analysis with three popular methods for choosing, including edgeR, DESeq2, and limma
- runMarker(): run biomarker identification to determine uniquely and significantly differential expressed genes for each subtype
- runGSEA(): run gene set enrichment analysis (GSEA), calculate activity of functional pathways and generate a pathway-specific heatmap
- runGSVA(): run gene set variation analysis to calculate enrichment score of each sample based on given gene set list of interest
- runNTP(): run nearest template prediction based on identified biomarkers to evaluate subtypes in external cohorts
- runPAM(): run partition around medoids classifier based on discovery cohort to predict subtypes in external cohorts
- runKappa(): run consistency evaluation using Kappa statistics between two appraisements that identify or predict current subtypes
该包已发表,使用时记得引用:
- Lu, X., Meng, J., Zhou, Y., Jiang, L., and Yan, F. (2020). MOVICS: an R package for multi-omics integration and visualization in cancer subtyping. bioRxiv, 2020.2009.2015.297820. [doi.org/10.1101/2020.09.15.297820]
安装
目前该包在github,只能通过以下方式安装,注意安装时最好先安装依赖包,因为这个包的依赖包非常多,安装过程中非常容易失败。对于初学者来说,这个包的安装不是很友好哦~
# 网络安装
devtools::install_github("xlucpu/MOVICS")
# 或者下载到本地安装
devtools::install_local("E:/R/R包/MOVICS-master.zip")
GET Module
准备数据
我们先看一下示例数据。
library(MOVICS)
##
使用该包自带数据进行演示,这个自带数据是已经清洗好的。过几天再专门写一篇推文介绍怎么准备这个数据。
# TCGA的乳腺癌数据
load(system.file("extdata", "brca.tcga.RData", package = "MOVICS", mustWork = TRUE))
load(system.file("extdata", "brca.yau.RData", package = "MOVICS", mustWork = TRUE))
brca.tcga里面是多个组学的数据,比如mRNA、lncRNA、甲基化、突变数据等,还有临床信息,比如生存时间和生存状态以及乳腺癌的PAM50分类。
为了演示,这个数据通过MAD筛选了部分数据:
- 500 mRNAs,
- 500 lncRNA,
- 1,000 promoter CGI probes/genes with high variation
- 30 genes that mutated in at least 3% of the entire cohort.
注意,这里最重要的一点是:每种组学的数据的样本数量、名字、顺序应该完全一致。大家可以自己看一下这些数据是什么样的。
names(brca.tcga)
## [1] "mRNA.expr" "lncRNA.expr" "meth.beta" "mut.status" "count"
## [6] "fpkm" "maf" "segment" "clin.info"
names(brca.yau)
## [1] "mRNA.expr" "clin.info"
# 提取"mRNA.expr""lncRNA.expr""meth.beta""mut.status"
mo.data <- brca.tcga[1:4]
# 提取raw count data
count <- brca.tcga$count
# 提取fpkm data
fpkm <- brca.tcga$fpkm
# 提取maf
maf <- brca.tcga$maf
# 提取segmented copy number
segment <- brca.tcga$segment
# 提取生存信息
surv.info <- brca.tcga$clin.info
筛选基因(降维)
getElites,顾名思义,找出精英,找出最牛逼的,也就是说这个函数可以做一些预处理和筛选工作,可以帮你进行数据准备工作。
主要可以做以下预处理:
- 缺失值插补:直接删除或者knn插补
- 筛选分子:可根据mad, sd, pca, cox, freq(二分类数据)进行筛选
其实这个不是第一步,第一步应该是自己先清洗一下数据,比如表达矩阵先进行log转换等。
下面是一些功能演示,还是非常强大的。
缺失值插补:
# scenario 1: 处理缺失值
tmp <- brca.tcga$mRNA.expr # get expression data
dim(tmp) # check data dimension
## [1] 500 643
tmp[1,1] <- tmp[2,2] <- NA # 添加几个NA
tmp[1:3,1:3] # check data
## BRCA-A03L-01A BRCA-A04R-01A BRCA-A075-01A
## SCGB2A2 NA 1.42 7.24
## SCGB1D2 10.11 NA 5.88
## PIP 4.54 2.59 4.35
elite.tmp <- getElites(dat = tmp,
method = "mad",
na.action = "rm", # 直接删除
elite.pct = 1) # 保留100%的数据
## --2 features with NA values are removed.
## missing elite.num then use elite.pct
dim(elite.tmp$elite.dat)
## [1] 498 643
elite.tmp <- getElites(dat = tmp,
method = "mad",
na.action = "impute", # 使用knn进行插补
elite.pct = 1)
## missing elite.num then use elite.pct
dim(elite.tmp$elite.dat)
## [1] 500 643
elite.tmp$elite.dat[1:3,1:3] # NA values have been imputed
## BRCA-A03L-01A BRCA-A04R-01A BRCA-A075-01A
## SCGB2A2 6.867 1.420 7.24
## SCGB1D2 10.110 4.739 5.88
## PIP 4.540 2.590 4.35
使用MAD筛选分子:
# scenario 2: 使用MAD筛选,最大中位差
tmp <- brca.tcga$mRNA.expr
elite.tmp <- getElites(dat = tmp,
method = "mad",
elite.pct = 0.1) # 保留MAD前10%的基因
## missing elite.num then use elite.pct
dim(elite.tmp$elite.dat) # 500的10%是50
## [1] 50 643
#> [1] 50 643
elite.tmp <- getElites(dat = tmp,
method = "sd",
elite.num = 100, # 保留MAD前100的基因
elite.pct = 0.1) # 此时这个参数就不起作用了
## elite.num has been provided then discards elite.pct.
dim(elite.tmp$elite.dat)
## [1] 100 643
使用PCA筛选分子,需要了解一些关于PCA的基础知识:R语言主成分分析
# scenario 3: 使用PCA筛选分子
tmp <- brca.tcga$mRNA.expr # get expression data with 500 features
elite.tmp <- getElites(dat = tmp,
method = "pca",
pca.ratio = 0.95) # 主成分的比例
## --the ratio used to select principal component is set as 0.95
dim(elite.tmp$elite.dat) # get 204 elite (PCs) left
## [1] 204 643
使用单因素COX回归筛选分子,也就是对每个分子做单因素cox分析,选择有意义的留下,需要提供生存信息:
# scenario 4: 使用cox筛选分子
tmp <- brca.tcga$mRNA.expr # get expression data
elite.tmp <- getElites(dat = tmp,
method = "cox",
surv.info = surv.info, # 生存信息,列名必须有'futime'和'fustat'
p.cutoff = 0.05,
elite.num = 100) # 此时这个参数也是不起作用的
## --all sample matched between omics matrix and survival data.
## 5% 10% 15% 20% 25% 30% 35% 40% 45% 50% 55% 60% 65% 70% 75% 80% 85% 90% 95% 100%
dim(elite.tmp$elite.dat) # get 125 elites
## [1] 125 643
table(elite.tmp$unicox$pvalue < 0.05) # 125 genes have nominal pvalue < 0.05 in
##
## FALSE TRUE
## 375 125
tmp <- brca.tcga$mut.status # get mutation data
elite.tmp <- getElites(dat = tmp,
method = "cox",
surv.info = surv.info,
p.cutoff = 0.05,
elite.num = 100)
## --all sample matched between omics matrix and survival data.
## 7% 13% 20% 27% 33% 40% 47% 53% 60% 67% 73% 80% 87% 93% 100%
dim(elite.tmp$elite.dat) # get 3 elites
## [1] 3 643
table(elite.tmp$unicox$pvalue < 0.05) # 3 mutations have nominal pvalue < 0.05
##
## FALSE TRUE
## 27 3
使用突变频率筛选分子,这个是准们用于0/1矩阵这种二分类数据的:
# scenario 5: 使用突变频率筛选
tmp <- brca.tcga$mut.status # get mutation data
rowSums(tmp)
## PIK3CA TP53 TTN CDH1 GATA3 MLL3 MUC16 MAP3K1 SYNE1 MUC12 DMD
## 208 186 111 83 58 49 48 38 33 32 31
## NCOR1 FLG PTEN RYR2 USH2A SPTA1 MAP2K4 MUC5B NEB SPEN MACF1
## 31 30 29 27 27 25 25 24 24 23 23
## RYR3 DST HUWE1 HMCN1 CSMD1 OBSCN APOB SYNE2
## 23 22 22 22 21 21 21 21
elite.tmp <- getElites(dat = tmp,
method = "freq", # must set as 'freq'
elite.num = 80, # 这里是指突变频率
elite.pct = 0.1) # 此时该参数不起作用
## --method of 'freq' only supports binary omics data (e.g., somatic mutation matrix), and in this manner, elite.pct and elite.num are used to cut frequency.
## elite.num has been provided then discards elite.pct.
rowSums(elite.tmp$elite.dat) # 只保留在80个及以上样本中突变的基因
## PIK3CA TP53 TTN CDH1
## 208 186 111 83
elite.tmp <- getElites(dat = tmp,
method = "freq",
elite.pct = 0.2)
## --method of 'freq' only supports binary omics data (e.g., somatic mutation matrix), and in this manner, elite.pct and elite.num are used to cut frequency.
## missing elite.num then use elite.pct
rowSums(elite.tmp$elite.dat) # only genes that are mutated in over than 0.2*643=128.6
## PIK3CA TP53
## 208 186
确定最佳亚型数量
根据分子表达量对样本进行分型,分子就是上一步得到的mRNA、lncRNA、miRNA、甲基化矩阵等。
先根据CPI和Gaps-statistics确定分成几个亚型:
optk.brca <- getClustNum(data = mo.data, # 4种组学数据
is.binary = c(F,F,F,T), #前3个不是二分类的,最后一个是
try.N.clust = 2:8, # 尝试亚型数量,从2到8
fig.name = "CLUSTER NUMBER OF TCGA-BRCA")#保存的文件名
## calculating Cluster Prediction Index...
## 5% complete
## 5% complete
## 10% complete
## 10% complete
## 15% complete
## 15% complete
## 20% complete
## 25% complete
## 25% complete
## 30% complete
## 30% complete
## 35% complete
## 35% complete
## 40% complete
## 45% complete
## 45% complete
## 50% complete
## 50% complete
## 55% complete
## 55% complete
## 60% complete
## 65% complete
## 65% complete
## 70% complete
## 70% complete
## 75% complete
## 75% complete
## 80% complete
## 85% complete
## 85% complete
## 90% complete
## 90% complete
## 95% complete
## 95% complete
## 100% complete
## calculating Gap-statistics...
## visualization done...
## --the imputed optimal cluster number is 3 arbitrarily, but it would be better referring to other priori knowledge.

会自动在当前工作目录下产生一个PDF格式的图片。
函数给出的结果是3,但是考虑到乳腺癌的PAM0分类,我们选择k=5,也就是分成5个亚型。
所以这个确定最佳亚型个数是根据你自己的需要来的哈,灵活调整~
根据单一算法分型
确定分成几个亚型之后,可以通过算法进行分型了。提供了非常多的方法,大家常见的非负矩阵分解、异质性聚类等等都提供了。
比如根据贝叶斯方法进行分型:
# perform iClusterBayes (may take a while)
iClusterBayes.res <- getiClusterBayes(data = mo.data,
N.clust = 5,
type = c("gaussian","gaussian","gaussian","binomial"),
n.burnin = 1800,
n.draw = 1200,
prior.gamma = c(0.5, 0.5, 0.5, 0.5),
sdev = 0.05,
thin = 3)
## clustering done...
## feature selection done...
或者使用统一的函数,自己选择方法即可,两种方法得到的结果完全是一样的:
iClusterBayes.res <- getMOIC(data = mo.data,
N.clust = 5,
methodslist = "iClusterBayes", # 指定算法
type = c("gaussian","gaussian","gaussian","binomial"), # data type corresponding to the list
n.burnin = 1800,
n.draw = 1200,
prior.gamma = c(0.5, 0.5, 0.5, 0.5),
sdev = 0.05,
thin = 3)
返回的结果包含一个clust.res对象,它有两列:clust列指示样本所属的亚型,samID列记录对应的样本名称。对于提供特征选择过程的算法(如iClusterBayes、CIMLR和MoCluster),结果还包含一个feat.res对象,存储了这种过程的信息。对于涉及分层聚类的算法(例如COCA、ConsensusClustering),样本聚类的相应树状图也将作为clust.dend返回,如果用户想要将它们放在热图中会很有用。
同时进行多种分型算法
可以同时根据多种算法进行分型,然后整合它们的结果,得到最终的结果,不是一般的强大:
# perform multi-omics integrative clustering with the rest of 9 algorithms
moic.res.list <- getMOIC(data = mo.data,
methodslist = list("SNF", "PINSPlus", "NEMO", "COCA", "LRAcluster", "ConsensusClustering", "IntNMF", "CIMLR", "MoCluster"), # 9种算法
N.clust = 5,
type = c("gaussian", "gaussian", "gaussian", "binomial"))
## --you choose more than 1 algorithm and all of them shall be run with parameters by default.
## SNF done...
## Clustering method: kmeans
## Perturbation method: noise
## PINSPlus done...
## NEMO done...
## COCA done...
## LRAcluster done...
## end fraction
## clustered
## ConsensusClustering done...
## IntNMF done...
## clustering done...
## feature selection done...
## CIMLR done...
## clustering done...
## feature selection done...
## MoCluster done...
再把贝叶斯的结果一起加进来,这就是10种算法了:
moic.res.list <- append(moic.res.list,
list("iClusterBayes" = iClusterBayes.res))
# 保存下结果
save(moic.res.list, file = "moic.res.list.rda")
整合多种分型结果
借鉴了consensus ensembles的想法,实现对多个分型算法结果的整合。
可以画出一个一致性热图:
load(file = "moic.res.list.rda")
cmoic.brca <- getConsensusMOIC(moic.res.list = moic.res.list,
fig.name = "CONSENSUS HEATMAP",
distance = "euclidean",
linkage = "average")

结果会保存在当前工作目录中。
查看分型结果的质量
除了通过上面的热图查看分型结果,还可以使用Silhouette准则判断分型质量。
以下是解释,来源于网络:
Silhouette准则是一种用于聚类分析中的评价方法,它通过对每个数据点与其所属簇内其他数据点之间的距离进行比较,来衡量聚类质量的好坏。Silhouette准则可以帮助我们确定最佳的聚类数量,从而提高聚类分析的可靠性和准确性。
Silhouette准则的计算方法如下:对于每个数据点i,计算它与同簇中其他数据点之间的平均距离ai,以及与最近其他簇中数据点之间的平均距离bi。然后,定义每个数据点的Silhouette系数为:
s(i) = (bi - ai) / max(ai, bi)
Silhouette系数的取值范围在-1到1之间,其中负值表示数据点更容易被分类到错误的簇中,而正值则表示数据点更容易被正确分类。Silhouette系数的平均值可以用来评估整个聚类的质量,因此,Silhouette准则的目标是最大化Silhouette系数的平均值,从而找到最佳的聚类数量。
当聚类数量增加时,Silhouette系数的平均值通常会先增加后减少。因此,我们需要找到一个聚类数量,使得Silhouette系数的平均值达到最大值。通常,我们会通过绘制Silhouette图来选择最佳的聚类数量。Silhouette图是一种以Silhouette系数为纵轴,聚类数量为横轴的图表,它可以帮助我们直观地理解聚类的质量。
在使用Silhouette准则进行聚类分析时,需要注意以下几点:
- Silhouette系数只适用于欧氏距离或相关度量,对于其他距离度量可能不适用。
- Silhouette系数的计算时间较长,因此在处理大规模数据时需要注意计算效率。
- Silhouette系数并不是唯一的评价指标,对于特定的聚类问题可能需要采用其他评价指标。
结果会保存在当前工作目录中:
getSilhouette(sil = cmoic.brca$sil, # a sil object returned by getConsensusMOIC()
fig.path = getwd(),
fig.name = "SILHOUETTE",
height = 5.5,
width = 5)

## png
## 2
多组学分型热图
分型之后,肯定是要对每个组学数据进行热图展示不同亚型的表达量情况。
不过需要做一些准备工作。
- 把甲基化的β值矩阵转换为M值矩阵,作者推荐,这样做展示效果更好;
- 数据标准化,画热图之钱一般都会进行这个操作,其实是通过
scale进行的,比如把所有数据压缩为[-2,2],超过2的用2表示,小于-2的用-2表示
# β值矩阵转换为M值矩阵
indata <- mo.data
indata$meth.beta <- log2(indata$meth.beta / (1 - indata$meth.beta))
# 对数据进行标准化
plotdata <- getStdiz(data = indata,
halfwidth = c(2,2,2,NA), # no truncation for mutation
centerFlag = c(T,T,T,F), # no center for mutation
scaleFlag = c(T,T,T,F)) # no scale for mutation
我们这里就用贝叶斯分型的结果进行展示,首先是提取每个组学的结果,然后每个组学中选择前10个分子进行标注:
feat <- iClusterBayes.res$feat.res
feat1 <- feat[which(feat$dataset == "mRNA.expr"),][1:10,"feature"]
feat2 <- feat[which(feat$dataset == "lncRNA.expr"),][1:10,"feature"]
feat3 <- feat[which(feat$dataset == "meth.beta"),][1:10,"feature"]
feat4 <- feat[which(feat$dataset == "mut.status"),][1:10,"feature"]
annRow <- list(feat1, feat2, feat3, feat4)
下面就是画图即可,其实也是借助complexheatmap实现的,只不过帮你简化了很多过程,结果会自动保存在当前工作目录下,MOVICS的默认出图还是很美观的,可能比你自己画的好看~
# 为每个组学的热图自定义颜色,不定义也可
mRNA.col <- c("#00FF00", "#008000", "#000000", "#800000", "#FF0000")
lncRNA.col <- c("#6699CC", "white" , "#FF3C38")
meth.col <- c("#0074FE", "#96EBF9", "#FEE900", "#F00003")
mut.col <- c("grey90" , "black")
col.list <- list(mRNA.col, lncRNA.col, meth.col, mut.col)
# comprehensive heatmap (may take a while)
getMoHeatmap(data = plotdata,
row.title = c("mRNA","lncRNA","Methylation","Mutation"),
is.binary = c(F,F,F,T), # the 4th data is mutation which is binary
legend.name = c("mRNA.FPKM","lncRNA.FPKM","M value","Mutated"),
clust.res = iClusterBayes.res$clust.res, # cluster results
clust.dend = NULL, # no dendrogram
show.rownames = c(F,F,F,F), # specify for each omics data
show.colnames = FALSE, # show no sample names
annRow = annRow, # mark selected features
color = col.list,
annCol = NULL, # no annotation for samples
annColors = NULL, # no annotation color
width = 10, # width of each subheatmap
height = 5, # height of each subheatmap
fig.name = "COMPREHENSIVE HEATMAP OF ICLUSTERBAYES")

上面是贝叶斯方法分型结果的展示,你也可以任选一种,毕竟我们有10种算法。
比如选择COCA法的结果进行展示,也是一模一样的用法,结果会自动保存:
# comprehensive heatmap (may take a while)
getMoHeatmap(data = plotdata,
row.title = c("mRNA","lncRNA","Methylation","Mutation"),
is.binary = c(F,F,F,T), # the 4th data is mutation which is binary
legend.name = c("mRNA.FPKM","lncRNA.FPKM","M value","Mutated"),
clust.res = moic.res.list$COCA$clust.res, # cluster results
clust.dend = moic.res.list$COCA$clust.dend, # show dendrogram for samples
color = col.list,
width = 10, # width of each subheatmap
height = 5, # height of each subheatmap
fig.name = "COMPREHENSIVE HEATMAP OF COCA")

如果你要展示多个临床信息,也是直接添加即可,注意自定义颜色需要使用circlize实现:
# extract PAM50, pathologic stage and age for sample annotation
annCol <- surv.info[,c("PAM50", "pstage", "age"), drop = FALSE]
# generate corresponding colors for sample annotation
annColors <- list(age = circlize::colorRamp2(breaks = c(min(annCol$age),
median(annCol$age),
max(annCol$age)),
colors = c("#0000AA", "#555555", "#AAAA00")),
PAM50 = c("Basal" = "blue",
"Her2" = "red",
"LumA" = "yellow",
"LumB" = "green",
"Normal" = "black"),
pstage = c("T1" = "green",
"T2" = "blue",
"T3" = "red",
"T4" = "yellow",
"TX" = "black"))
# comprehensive heatmap (may take a while)
getMoHeatmap(data = plotdata,
row.title = c("mRNA","lncRNA","Methylation","Mutation"),
is.binary = c(F,F,F,T), # the 4th data is mutation which is binary
legend.name = c("mRNA.FPKM","lncRNA.FPKM","M value","Mutated"),
clust.res = cmoic.brca$clust.res, # consensusMOIC results
clust.dend = NULL, # show no dendrogram for samples
show.rownames = c(F,F,F,F), # specify for each omics data
show.colnames = FALSE, # show no sample names
show.row.dend = c(F,F,F,F), # show no dendrogram for features
annRow = NULL, # no selected features
color = col.list,
annCol = annCol, # annotation for samples
annColors = annColors, # annotation color
width = 10, # width of each subheatmap
height = 5, # height of each subheatmap
fig.name = "COMPREHENSIVE HEATMAP OF CONSENSUSMOIC")

是不是非常牛逼?
到这里第一部分的内容就介绍完了,下面就是探索、比较不同的亚型了。

开放原子开发者工作坊旨在鼓励更多人参与开源活动,与志同道合的开发者们相互交流开发经验、分享开发心得、获取前沿技术趋势。工作坊有多种形式的开发者活动,如meetup、训练营等,主打技术交流,干货满满,真诚地邀请各位开发者共同参与!
更多推荐
 21
21 0
0- 0



 已为社区贡献38条内容
已为社区贡献38条内容

所有评论(0)