GSVA和ssGSEA
ORA和GSEA。假如你手上有一撮基因,但是你不知道它们有哪些功能,你可以先做个ORA富集分析;假如你有一撮基因,你想看看它们在两种状态下分别会富集在哪些通路,或者两种状态下的功能会有哪些不一样,那你可以做GSEA。平常最常见的GO和KEGG只是已知功能的基因集合而已,这些基因的功能我们已经研究透了,现在把它们放一起,用来方便大家探索你手上的基因可能有哪些功能,这就是注释基因集,用已知功能的基因来
最近写了这么多关于富集分析的推文,不知道大家看懂了没有,其实富集分析主要就分为两种:ORA和GSEA。
假如你手上有一撮基因,但是你不知道它们有哪些功能,你可以先做个ORA富集分析;假如你有一撮基因,你想看看它们在两种状态下分别会富集在哪些通路,或者两种状态下的功能会有哪些不一样,那你可以做GSEA。
平常最常见的GO和KEGG只是已知功能的基因集合而已,这些基因的功能我们已经研究透了,现在把它们放一起,用来方便大家探索你手上的基因可能有哪些功能,这就是注释基因集,用已知功能的基因来注释你手上的基因。
除了GO和KEGG,还有非常多的注释基因集,比如我们之前介绍过的WikiPathways、Reactome等等。
GSVA是GSEA的变种方法,它是一种常见的可以为样本打分的方法,可以把行为基因列为样本的表达矩阵变为行为基因集列为样本的表达矩阵,也就是说,你提供一个行为基因列为样本的表达矩阵以及几个注释基因集,它就可以计算出样本的变异分数,返回一个每行是一个基因集,列为样本的矩阵。
网上常见的根据通路对样本打分的方法说的就是这个GSVA。
ssGSEA是GSVA的一种特殊类型,二者没有本质上的区别,除了这两种,还有zscore和plage方法,都是通过GSVA包实现的。
我们使用TCGA-SKCM的数据进行演示,注释基因集一般是从misigdb网站下载的,根据你自己的需求来,有些人想看看免疫相关的,那你就下载免疫相关的基因集,你想看炎症相关的就下载炎症相关的基因集。
准备基因集
我们就从msigdb下载经典的Hallmark_gene_sets。下载之后使用clusterProfiler的read.gmt函数直接读取,然后使用split变成GSVA需要的格式。
hall_mark <- "G:/bioinfo/000files/h.all.v2023.1.Hs.symbols.gmt"
# 结果是一个data.frame
genesets <- clusterProfiler::read.gmt(hall_mark)
##
head(genesets)
## term gene
## 1 HALLMARK_TNFA_SIGNALING_VIA_NFKB JUNB
## 2 HALLMARK_TNFA_SIGNALING_VIA_NFKB CXCL2
## 3 HALLMARK_TNFA_SIGNALING_VIA_NFKB ATF3
## 4 HALLMARK_TNFA_SIGNALING_VIA_NFKB NFKBIA
## 5 HALLMARK_TNFA_SIGNALING_VIA_NFKB TNFAIP3
## 6 HALLMARK_TNFA_SIGNALING_VIA_NFKB PTGS2
# 按照term对symbol进行分组,变成list
genesets4gsva <- split(genesets$gene, genesets$term)
class(genesets4gsva)
## [1] "list"
length(genesets4gsva)
## [1] 50
#genesets4gsva[1:4]
这个split的操作在之前的泛癌可视化中也介绍过:任意基因在泛癌中的表达量可视化
大家可以自己尝试下看看具体的格式,这个格式在免疫浸润分析中也用过的:
准备表达矩阵
我们从TCGA下载黑色素瘤的转录组数据,使用easyTCGA,1行代码解决,即可得到6种表达矩阵和临床信息,而且是官网最新的数据:
library(easyTCGA)
getmrnaexpr("TCGA-SKCM")
加载数据:
load(file = "G:/easyTCGA_test/output_mRNA_lncRNA_expr/TCGA-SKCM_mrna_expr_tpm.rdata")
这个数据是直接从GDC的官网数据中提取出来的,没有经过任何转化,所以我们先进行log2转换。
expr <- log2(mrna_expr_tpm+1)
dim(expr)
## [1] 19938 473
expr[1:4,1:4]
## TCGA-EB-A3Y6-01A-21R-A239-07 TCGA-D9-A4Z6-06A-12R-A266-07
## MT-CO2 15.82250 15.25351
## MT-CO3 15.38751 14.93694
## MT-ND4 14.67998 15.34512
## MT-CO1 15.22099 15.14673
## TCGA-FW-A5DY-06A-11R-A311-07 TCGA-EE-A2GH-06A-11R-A18T-07
## MT-CO2 16.32066 14.97308
## MT-CO3 15.90499 14.58077
## MT-ND4 15.67466 14.42920
## MT-CO1 16.02932 14.58028
一共有19938个mRNA和473个样本。
GSVA分析
下面就开始进行GSVA分析了,代码其实非常简单:
library(GSVA)
expr_geneset <- gsva(expr = as.matrix(expr), # 不能是data.frame
gset.idx.list = genesets4gsva,
method="gsva",
kcdf="Gaussian", # log后的tpm用高斯分布
parallel.sz=10 # 多线程
)
## Setting parallel calculations through a MulticoreParam back-end
## with workers=10 and tasks=100.
## Estimating GSVA scores for 50 gene sets.
## Estimating ECDFs with Gaussian kernels
## Estimating ECDFs in parallel on 10 cores
##
|
| | 0%
|======================================================================| 100%
dim(expr_geneset)
## [1] 50 473
expr_geneset[1:4,1:2]
## TCGA-EB-A3Y6-01A-21R-A239-07
## HALLMARK_TNFA_SIGNALING_VIA_NFKB -0.30185257
## HALLMARK_HYPOXIA -0.19158522
## HALLMARK_CHOLESTEROL_HOMEOSTASIS -0.01329616
## HALLMARK_MITOTIC_SPINDLE -0.28850995
## TCGA-D9-A4Z6-06A-12R-A266-07
## HALLMARK_TNFA_SIGNALING_VIA_NFKB -0.15484044
## HALLMARK_HYPOXIA -0.07166491
## HALLMARK_CHOLESTEROL_HOMEOSTASIS -0.39672253
## HALLMARK_MITOTIC_SPINDLE -0.10932485
# 结果是matrix,变成data.frame方便使用
expr_geneset <- as.data.frame(expr_geneset)
Hallmark中只有50个数据集,而且我们用了10个线程,所以这里还是蛮快的。
结果是50行,对应着我们的50个基因集,473列,依然是对应着473个样本。
这个结果和我们的原始表达矩阵有区别吗?没有
所以对原始表达矩阵可以做的操作都可以对这个expr_geneset做,比如差异分析,生存分析等等。
后续分析
有了这个结果,我们就可以做很多事情,因为它本质上也是一个表达矩阵而已。比如我想看看某个基因和炎症反应的关系,这有何难?做个相关性分析不就行了吗?
我们就以HOPX这个基因为例。
首先提取下这个HOPX和炎症反应的表达矩阵:
HOPX_expr <- expr["HOPX",]
HOPX_expr[,1:4]
## TCGA-EB-A3Y6-01A-21R-A239-07 TCGA-D9-A4Z6-06A-12R-A266-07
## HOPX 1.198746 0.4733194
## TCGA-FW-A5DY-06A-11R-A311-07 TCGA-EE-A2GH-06A-11R-A18T-07
## HOPX 1.760987 2.010207
inflam_expr <- expr_geneset["HALLMARK_INFLAMMATORY_RESPONSE",]
inflam_expr[1:4,1:4]
## TCGA-EB-A3Y6-01A-21R-A239-07
## HALLMARK_INFLAMMATORY_RESPONSE -0.2640837
## NA NA
## NA.1 NA
## NA.2 NA
## TCGA-D9-A4Z6-06A-12R-A266-07
## HALLMARK_INFLAMMATORY_RESPONSE -0.4386833
## NA NA
## NA.1 NA
## NA.2 NA
## TCGA-FW-A5DY-06A-11R-A311-07
## HALLMARK_INFLAMMATORY_RESPONSE 0.2886898
## NA NA
## NA.1 NA
## NA.2 NA
## TCGA-EE-A2GH-06A-11R-A18T-07
## HALLMARK_INFLAMMATORY_RESPONSE 0.4181877
## NA NA
## NA.1 NA
## NA.2 NA
然后就是计算HPOX和凋亡通路的相关性和P值:
identical(colnames(HOPX_expr),colnames(inflam_expr))
## [1] TRUE
cor.test(t(HOPX_expr),t(inflam_expr))
##
## Pearson's product-moment correlation
##
## data: t(HOPX_expr) and t(inflam_expr)
## t = 9.3453, df = 471, p-value < 2.2e-16
## alternative hypothesis: true correlation is not equal to 0
## 95 percent confidence interval:
## 0.3166304 0.4689398
## sample estimates:
## cor
## 0.3955007
你还想再画个图?那真是轻而易举,我们画个简单的,下面这张图我们在免疫浸润可视化中也用过,而且是批量出图的:免疫浸润可视化
library(ggplot2)
library(ggpubr)
plot_df <- data.frame(t(HOPX_expr), t(inflam_expr))
names(plot_df) <- c("hopx","inflam")
head(plot_df)
## hopx inflam
## TCGA-EB-A3Y6-01A-21R-A239-07 1.1987456 -0.2640837
## TCGA-D9-A4Z6-06A-12R-A266-07 0.4733194 -0.4386833
## TCGA-FW-A5DY-06A-11R-A311-07 1.7609873 0.2886898
## TCGA-EE-A2GH-06A-11R-A18T-07 2.0102069 0.4181877
## TCGA-EE-A2GR-06A-11R-A18S-07 0.2441564 -0.5117789
## TCGA-EB-A4XL-01A-11R-A27Q-07 5.7113490 0.1410712
ggplot(plot_df, aes(hopx, inflam))+
geom_point()+
geom_rug()+
geom_smooth(method = "lm",color="blue")+
stat_cor(method = "spearman",color="red")
## `geom_smooth()` using formula = 'y ~ x'

如果你还想继续美化,那你需要自己学习ggplot2,还需要多看文献,模仿别人!
当然这个图还可以更加花里胡哨,使用我们之前介绍过的ggstatsplot:统计可视化的颜值天花板:ggstatsplot
library(ggstatsplot)
## You can cite this package as:
## Patil, I. (2021). Visualizations with statistical details: The 'ggstatsplot' approach.
## Journal of Open Source Software, 6(61), 3167, doi:10.21105/joss.03167
ggscatterstats(data = plot_df,
x = hopx,
y = inflam,
xlab = "log2(HOPX TPM + 1)",
ylab = "Inflammatory Response",
bf.message = F
)
## Registered S3 method overwritten by 'ggside':
## method from
## +.gg ggplot2
## `stat_bin()` using `bins = 30`. Pick better value with `binwidth`.
## `stat_bin()` using `bins = 30`. Pick better value with `binwidth`.

什么?你还想批量计算所有通路和HOPX的相关性,那就赶紧看我们之前介绍过的方法吧:单基因富集分析
一个小测试
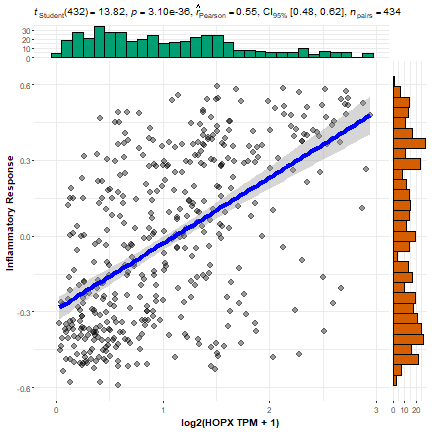
从图中可以看出这个相关性不是很好,只有0.4左右,我认为这是由于HOPX的表达矩阵里有一些异常样本(或者叫离群值吧),比如图的右侧有一些样本很离散的,离多数样本很远的。我们尝试下把这些离群值删除,再重新画图看看。
suppressMessages(library(dplyr))
plot_df %>%
filter(hopx < 3) %>%
ggscatterstats(data = .,
x = hopx,
y = inflam,
xlab = "log2(HOPX TPM + 1)",
ylab = "Inflammatory Response",
bf.message = F
)
## `stat_bin()` using `bins = 30`. Pick better value with `binwidth`.
## `stat_bin()` using `bins = 30`. Pick better value with `binwidth`.

果然!结果和我预想的一样,相关性从0.4提升到了0.55!对于很多结果来说这就是起死回生了!
但是,这种数据操作,你如果不能解释清楚,那算操纵数据吗?

开放原子开发者工作坊旨在鼓励更多人参与开源活动,与志同道合的开发者们相互交流开发经验、分享开发心得、获取前沿技术趋势。工作坊有多种形式的开发者活动,如meetup、训练营等,主打技术交流,干货满满,真诚地邀请各位开发者共同参与!
更多推荐
 1
1 0
0- 0



 已为社区贡献38条内容
已为社区贡献38条内容

所有评论(0)