locuszoomr!这个 R 包可视化 GWAS 结果数据绝对够顶
生信碱移locuszoomr可视化GWAS结果locuszoomr 是由伦敦玛丽王后大学的研究者开发的R包,允许用户生成出版级别的基因组位点注释图,这可以用于可视化并解读 GWAS 分析结果。由 locuszoomr 生成的基因位点注释图通过 R 基础图形或 ggplot2 生成,简易使用的同时又附加了大量个性化参数设置。本文将介绍locuszoomr的使用方法,大家也可以根据官方文档探索更多的个
生信碱移
locuszoomr可视化GWAS结果
locuszoomr 是由伦敦玛丽王后大学的研究者开发的R包,允许用户生成出版级别的基因组位点注释图,这可以用于可视化并解读 GWAS 分析结果。由 locuszoomr 生成的基因位点注释图通过 R 基础图形或 ggplot2 生成,简易使用的同时又附加了大量个性化参数设置。

本文将介绍locuszoomr的使用方法,大家也可以根据官方文档探索更多的个性化玩法:
-
https://github.com/harryyiheyang/EGG
1、locuszoomr的安装
用户可以通过以下代码从 gihub 安装 locuszoomr 及相关依赖包:
# 一些依赖包的安装
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("ensembldb")
BiocManager::install("EnsDb.Hsapiens.v75")
BiocManager::install("AnnotationFilter")
BiocManager::install("GenomicRanges")
BiocManager::install("rtracklayer")
# 从cran库安装
install.packages("locuszoomr")
2、示例数据加载
这里展示的是内置数据SLE_gwas_sub,其实也就是在 GWAS 的结果之上添加了一些其他想要可视化的信息(比如r2,这些添加的信息都是非必须的,大家只需要保证自己的数据有常规的GWAS的表头即可):
data(SLE_gwas_sub)
# 看一下这个数据的表头
head(SLE_gwas_sub)
# chrom pos rsid other_allele effect_allele p beta se OR
#1 2 191794580 rs193239665 A T 0.000723856 0.32930375 0.09741618 1.39
#2 2 191794978 rs72907256 C T 0.000481744 0.39877612 0.11423935 1.49
#3 2 191795546 rs6434429 C G 0.156723000 -0.09431068 0.06659515 0.91
#4 2 191795869 rs148265823 A G 0.606197000 -0.04082199 0.07918766 0.96
#5 2 191799600 rs60202309 T G 0.100580000 0.07696104 0.04686893 1.08
#6 2 191800180 rs114544034 T C 0.022496800 -0.16251893 0.07122170 0.85
# OR_lower OR_upper r2
#1 1.1483981 1.6824305 0.037
#2 1.1910878 1.8639264 0.034
#3 0.7986462 1.0368796 0.004
#4 0.8219877 1.1211846 0.004
#5 0.9852084 1.1839119 0.001
#6 0.7392542 0.9773364 0.019
表头的解释如下:
-
chrom:表示基因位点所在的染色体号;
-
pos:基因位点在染色体上的具体位置;
-
rsid:单核苷酸多态性(SNP)的参考编号;
-
other_allele:在该位点上的其他等位基因;
-
effect_allele:在该位点上与性状或疾病关联的等位基因;
-
p值:显著性水平,表示效应等位基因与性状或疾病关联的显著性;
-
beta:效应等位基因对性状或疾病的效应估计值,正值表示正相关,负值表示负相关;
-
se:效应估计值的标准误差,表示效应估计值的不确定性;
-
OR:效应等位基因的比值比,表示效应等位基因与性状或疾病关联的相对风险;
-
OR_lower:比值比的95%置信区间下限,表示比值比的最低估计值;
-
OR_upper:比值比的95%置信区间上限,表示比值比的最高估计值;
-
r2:这里是作者自己加的,与指定基因的连锁不平衡程度,大家可以自行加入自己感兴趣的数值。
3、使用演示
① 可视化指定基因区间的结构注释以及 gwas 显著值:
loc <- locus(data = SLE_gwas_sub, # 内置函数detect_cols会自动检测
gene = 'UBE2L3',
flank = 1e5, # 显示指定基因位点上下游范围100kb个碱基
ens_db = "EnsDb.Hsapiens.v75")
summary(loc)
locus_plot(loc)

注意,locus的内置函数detect_cols会自动检测输入数据的表头,找到符合GWAS规律的数值。如果你不放心,也可以通过以下参数指定:
-
chrom:确定数据中包含染色体信息的列,如果为NULL则尝试自动检测该列; -
pos:确定数据中包含位置信息的列,如果为NULL则尝试自动检测该列; -
p:确定数据中包含SNP p值的列,如果为NULL则尝试自动检测该列; -
labs:确定数据中包含SNP rs ID的列,如果为NULL则尝试自动检测该列; -
index_snp:指定索引SNP,如果未指定,则选择p值最低的SNP,可以用于指定基因座区域而不是指定gene、seqname和xrange; -
LD:可选的字符值,指定数据中包含LD信息的列。
不仅如此,可以通过locus函数中的flank参数指定绘制范围:
loc <- locus(data = SLE_gwas_sub,
gene = 'UBE2L3',
flank = c(1e4, 1e5), # 分别指定上下游范围10kb与100kb
ens_db = "EnsDb.Hsapiens.v75")
summary(loc)
locus_plot(loc)

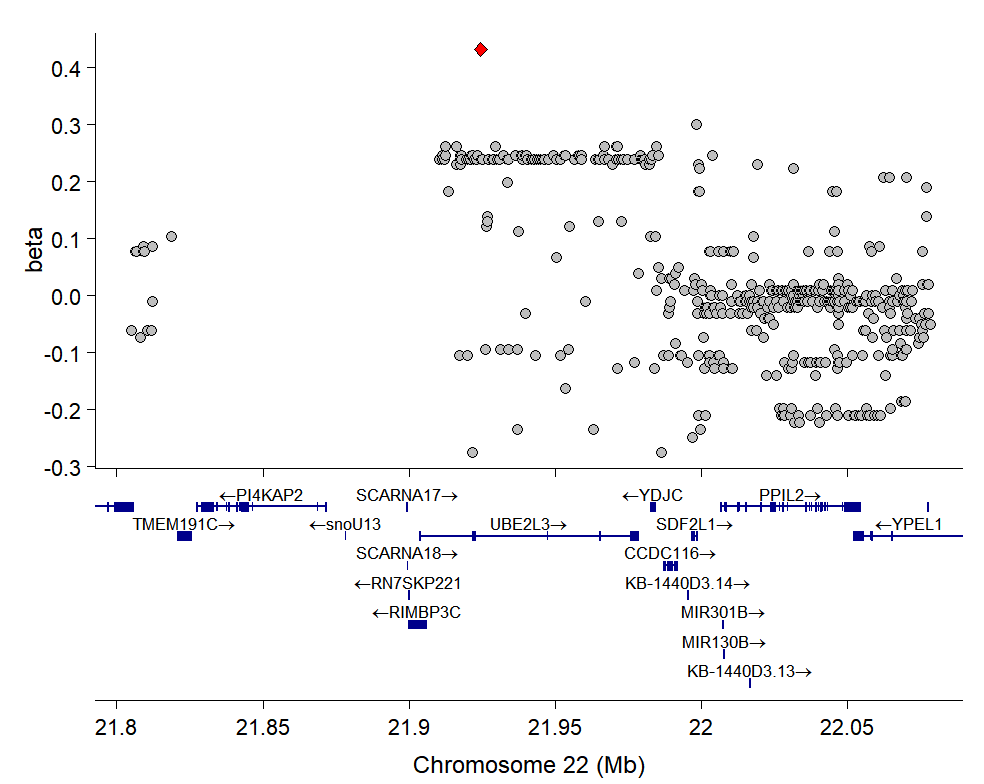
如果你的数据表还有其他的列,也可以在locus函数中使用yvar更改展示的y轴:
locb <- locus(SLE_gwas_sub,
gene = 'UBE2L3',
flank = 1e5,
yvar = "beta", # y轴展示为beta
ens_db = "EnsDb.Hsapiens.v75")
locus_plot(locb)

除了修改locus函数以外,locus_plot函数也具有多种参数用于个性化绘图,比如可以使用labels与label_x参数对指定的snp进行标记:
# 5.添加标注
loc <- locus(SLE_gwas_sub,
gene = 'UBE2L3',
flank = 1e5,
ens_db = "EnsDb.Hsapiens.v75")
locus_plot(loc,
labels = c("index", "rs140492"), # 标注指定的snp
label_x = c(4, -5)) # 标注的label在图上的x轴位置

除了标记 snp 以外,locus_plot函数也支持以下参数对注释轨迹进行筛选与高亮:
loc <- locus(data = SLE_gwas_sub,
gene = 'UBE2L3',
ens_db = "EnsDb.Hsapiens.v75")
summary(loc)
# ① 仅展示基因注释中的蛋白质编码基因
locus_plot(loc,
filter_gene_biotype = "protein_coding")
# ② 仅展示指定的基因注释
locus_plot(loc, filter_gene_name = c('UBE2L3', 'RIMBP3C', 'YDJC', 'PPIL2',
'PI4KAP2', 'MIR301B'))
# ③ 高亮指定基因
locus_plot(loc,
highlight = 'PPIL2')这里就不展示图片了,更多个性化的设置可以参考官方文档。
② 该软件可以使用LDlink api调用在线数据库的r²,用于可视化连锁不平衡的强度。使用以下代码即可显示区域内所有SNPs与指定SNP之间的r²水平(r²值用于量化两个SNP在遗传上的相关性,用于衡量连锁不平衡):
loc <- locus(SLE_gwas_sub,
gene = 'UBE2L3',
flank = 1e5,
ens_db = "EnsDb.Hsapiens.v75",
index_snp="rs140492") # 指定索引snp,如果不输入这个参数,则为p最小的snp
loc <- link_LD(loc, token = "这里输入你的token") # 获取LDlink数据,api提交一下邮箱信息就可以获得:https://ldlink.nih.gov/?tab=apiaccess
locus_plot(loc)

这里需要输入token,获得的方式很简单,在下方网页提交以下邮箱信息即可:
-
https://ldlink.nih.gov/?tab=apiaccess
此外需要注意,如果不指定SNP,那默认选择的 SNP 将会是 p 值最低的 SNP(index_snp参数用于手动指定SNP)。
③ 调用LDlink api也可以获得GTEX数据库的组织eqtl数据。以下代码在基因区间内同时可视化GWAS与eqtl结果(同样需要输入上面提到的 token):
# 注意看下面的注释哟
loc2 <- locus(SLE_gwas_sub,
gene = 'IRF5',
flank = c(7e4, 2e5),
ens_db = "EnsDb.Hsapiens.v75")
loc2 <- link_eqtl(loc2,
token = "此处输入你的token") # 同上,输入你的token
# 展示不同组织拥有的指定区间基因相关的eqtl数目
as.data.frame( table(loc2$LDexp$Tissue) )
# Var1 Freq
#1 Adipose - Subcutaneous 78
#2 Adipose - Visceral (Omentum) 69
#3 Adrenal Gland 38
#4 Artery - Aorta 109
#5 Artery - Coronary 11
#6 Artery - Tibial 112
#7 Brain - Cerebellar Hemisphere 5
#8 Brain - Putamen (basal ganglia) 32
#9 Breast - Mammary Tissue 38
#10 Cells - Cultured fibroblasts 122
#11 Colon - Sigmoid 40
#12 Colon - Transverse 60
#13 Esophagus - Gastroesophageal Junction 38
#14 Esophagus - Mucosa 92
#15 Esophagus - Muscularis 39
#16 Heart - Atrial Appendage 38
#17 Heart - Left Ventricle 40
#18 Liver 64
#19 Lung 137
#20 Muscle - Skeletal 38
#21 Nerve - Tibial 141
#22 Pancreas 98
#23 Pituitary 37
#24 Prostate 35
#25 Skin - Not Sun Exposed (Suprapubic) 70
#26 Skin - Sun Exposed (Lower leg) 85
#27 Small Intestine - Terminal Ileum 101
#28 Spleen 89
#29 Stomach 38
#30 Testis 219
#31 Thyroid 114
#32 Whole Blood 337
# 附加指定组织的eqtl结果
overlay_plot(loc2,
eqtl_gene = "IRF5", # 指定基因的eqtl
tissue = "Whole Blood") # 指定组织

④ 添加遗传重组率:
loc3 <- locus(SLE_gwas_sub,
gene = 'STAT4',
flank = 1e5,
ens_db = "EnsDb.Hsapiens.v75")
loc3 <- link_recomb(loc3,
genome = "hg19")
locus_plot(loc3)

⑤ 可视化基因结构注释,通过genetracks函数中的多种参数可以个性化基因轨迹图:
loc <- locus(SLE_gwas_sub,
gene = 'UBE2L3',
flank = 1e5,
ens_db = "EnsDb.Hsapiens.v75")
genetracks(loc, maxrows = 3,
filter_gene_biotype = 'protein_coding',
gene_col = 'grey', # 整个基因的颜色
exon_col = 'orange', # 外显子的颜色
exon_border = 'darkgrey', # 外显子边界的颜色
highlight='UBE2L3', # 指定高亮的基因
highlight_col = "red") # 高亮基因的颜色

⑥ 通过multi_layout函数进行多图排列:
loc1 <- locus(SLE_gwas_sub,
gene = 'IRF5',
flank = c(7e4, 2e5),
LD = "r2",
ens_db = "EnsDb.Hsapiens.v75")
loc2 <- locus(SLE_gwas_sub,
gene = 'STAT4',
flank = 1e5,
LD = "r2",
ens_db = "EnsDb.Hsapiens.v75")
multi_layout(ncol = 2,
plots = {
locus_plot(loc1, use_layout = FALSE, legend_pos = 'topleft')
locus_plot(loc2, use_layout = FALSE, legend_pos = NULL)
})

⑦ 分区块叠加绘制:
# 分区块绘制,添加更多的绘图信息
loc <- locus(SLE_gwas_sub,
gene = 'UBE2L3',
flank = 1e5,
ens_db = "EnsDb.Hsapiens.v75")
oldpar <- set_layers(2) # 基础图形之上叠加的数量
scatter_plot(loc, xticks = FALSE) # 基础的图形
line_plot(loc, # 叠加图1
col = "orange",
xticks = FALSE)
genetracks(loc) # 叠加图2
par(oldpar) # revert par() settings

⑧ 访问ensembl的在线基因组注释:
library(AnnotationHub)
ah <- AnnotationHub()
query(ah, c("EnsDb", "Homo sapiens")) # 查询人的注释数据
## AnnotationHub with 25 records
## # snapshotDate(): 2023-04-25
## # $dataprovider: Ensembl
## # $species: Homo sapiens
## # $rdataclass: EnsDb
## # additional mcols(): taxonomyid, genome, description, coordinate_1_based, maintainer,
## # rdatadateadded, preparerclass, tags, rdatapath, sourceurl, sourcetype
## # retrieve records with, e.g., 'object[["AH53211"]]'
##
## title
## AH53211 | Ensembl 87 EnsDb for Homo Sapiens
## ... ...
## AH100643 | Ensembl 106 EnsDb for Homo sapiens
## AH104864 | Ensembl 107 EnsDb for Homo sapiens
## AH109336 | Ensembl 108 EnsDb for Homo sapiens
## AH109606 | Ensembl 109 EnsDb for Homo sapiens
## AH113665 | Ensembl 110 EnsDb for Homo sapiens
ensDb_v106 <- ah[["AH100643"]] # 针对106这个版本,输入其编号AH100643进行查询
# built-in mini dataset
data("SLE_gwas_sub")
loc <- locus(data = SLE_gwas_sub,
gene = 'UBE2L3',
fix_window = 1e6,
ens_db = ensDb_v106) # 使用更新的注释数据绘图
locus_plot(loc)⑨ locus_ggplot、gg_genetracks、gg_sca函数 提供 ggplot2 框架的可视化方案:
locus_ggplot(loc)
gg_genetracks(loc)
gg_scatter(loc)就分享到这里
欢迎各位老铁关注

开放原子开发者工作坊旨在鼓励更多人参与开源活动,与志同道合的开发者们相互交流开发经验、分享开发心得、获取前沿技术趋势。工作坊有多种形式的开发者活动,如meetup、训练营等,主打技术交流,干货满满,真诚地邀请各位开发者共同参与!
更多推荐
 21
21 0
0- 0



 已为社区贡献4条内容
已为社区贡献4条内容

所有评论(0)