【生信】初探蛋白质性质和结构分析
对蛋白质的一级、二级和三级结构进行分析和预测、蛋白质的跨膜结构、翻译后修饰、亚细胞定位等进行分析和预测
实验目的
- 熟悉蛋白质序列和结构的主要分析内容
- 在实践中逐步理解蛋白质序列和结构的主要分析算法的基本原理
实验内容
- 综合使用多种在线工具,对蛋白质的一级、二级和三级结构进行分析和预测
- 综合使用多种在线工具,对蛋白质的跨膜结构、翻译后修饰、亚细胞定位等进行分析和预测
实验题目
第一题:Nanog分析
Nanog是2003年5月发现的一种转录因子,是一个有助于胚胎干细胞自我更新的关键因子,被认为在胚胎干细胞的全能性维持中起关键作用。 人源Nanog基因,通常写成Nanog1,位于12号染色体上。针对该基因(AY230262),请完成以下分析:
- 预测该基因编码产物的亚细胞定位。(至少使用两种预测方法,并比较不同方法的预测结果是否一致)
- 人Nanog基因产物是否是糖蛋白?什么类型的糖蛋白?
- 分析人Nanog基因产物的亲水性和疏水性,列出最亲水和最疏水的位点。
第二题:甲虫基因编码的蛋白质分析
分析一个甲虫基因(AF422804)编码的蛋白质的化学性质和结构特点(请注明分析方法名称):
- 等电点是多少?分子量是多少?是否含有pfam保守结构域?如有,列出登录号。
- 是否膜蛋白质?如果是膜蛋白质,请注明跨膜结构位点。
- 是否具有GPI固定(anchor)的蛋白质?
- 预测该蛋白序列的二级结构,并简述预测结果(至少使用两种预测方法,并比较不同方法的预测结果是否一致);
- 使用Swiss-Model 预测该蛋白质序列的三级结构,并简述预测结果;该蛋白在AlphaFold DB中是否有预测结果?如有,简述预测结果。
实验过程
Nanog分析
预测亚细胞定位
下载蛋白质序列
首先在ncbi上面检索AY230262这个基因,然后下载其编码的蛋白质的序列信息。


WoLF PSORT
WoLF PSORT是用于蛋白质亚细胞定位预测的PSORT II程序的扩展。WoLF PSORT将蛋白质氨基酸序列转化为数值定位特征。转换特征后,使用简单的k近邻分类器进行预测。在网站上,每个预测的证据以两种方式显示:与查询的定位特征最相似的已知定位蛋白质列表,以及关于个别定位特征的详细信息的表格。而且为了方便起见,还提供了查询到相似蛋白质的序列比对以及到UniProt和Gene Ontology的链接。
网站在这:https://wolfpsort.hgc.jp/

从下图的预测结果可以看到,Nanog最有可能位于细胞核。这个结果有三个部分:
第一行是预测结果的总结。亚细胞定位被缩写为4个字符,带有下划线的就是双重定位,后面的数字大致可以认为是查询序列的最近邻
下面这个表是KNN算法的邻居列表,显示了与查询蛋白质相似的蛋白质列表的一部分。对于每个邻居,如下所示:UniProt ID、定位位置、定位特征与查询序列之间的距离、占查询序列的百分比(the percent identity to the query)、其UniProt的链接、来自UniProt的亚细胞定位以及其他可用的定位信息。
再下面是标准化后的特征表。这些值被标准化为相对于WoLF PSORT训练数据的百分位数。蓝色显示的邻居值与查询值的差在10%以内,而红色显示的邻居值与查询值的差在20或更多。
Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007 Jul;35(Web Server issue):W585-7. doi: 10.1093/nar/gkm259. Epub 2007 May 21. PMID: 17517783; PMCID: PMC1933216.


YLoc
YLoc是一种可解释的蛋白质亚细胞定位预测系统。除了预测的位置,YLoc给出了为什么做出这一预测的原因,以及蛋白质序列的哪些生物学特性导致了这一预测。此外,置信度估计有助于用户将预测评价为可信。YLoc+能够准确预测多靶标蛋白的位置。
Sebastian Briesemeister, J�rg Rahnenf�hrer, Oliver Kohlbacher, YLoc—an interpretable web server for predicting subcellular localization, Nucleic Acids Research, Volume 38, Issue suppl_2, 1 July 2010, Pages W497–W502,

从下面的结果可以看到,Nanog最有可能位于细胞核,和WoLF PSORT的结果一致。
第一个表格是显示了YLoc的预测各定位的概率。亚细胞位置按其概率排序,从最可能的位置开始。最可能的位置或位置组合用红色突出显示。
最相似的蛋白质是来自Swiss-Prot 42.0的蛋白质,与所查询的蛋白质具有最高的局部序列同一性。其中,与这些蛋白质相关的GO项被用于预测。然而,由于它是最相似的蛋白质,我们在这里再次列出它。

下表列出了这个特定YLoc预测最重要的特征,从对预测影响最大的属性开始。一个(双)加号表示属性(强烈)支持此本地化的决策,而一个(双)减号表示属性(强烈)支持反对此本地化的决策。将鼠标光标放在一个字段上,查看来自该定位的蛋白质与查询蛋白质具有相同特征值的比例。
常用缩写:Cy = ‘细胞质’,Mi = ‘线粒体’,Nu = ‘细胞核’,SP = ‘分泌通路’。

DeelLoc 2.0
DeepLoc 2.0预测真核生物蛋白质的亚细胞定位。DeepLoc 2.0是一个多标签预测器,这意味着能够预测任何给定蛋白质的一个或多个定位。它可以区分10个不同的定位:细胞核,细胞质,细胞外,线粒体,细胞膜,内质网,叶绿体,高尔基体,溶酶体/液泡和过氧化物酶体。此外,DeepLoc 2.0可以预测对亚细胞定位预测有影响的排序信号的存在。
DeepLoc 2.0相对于1.0整体模型性能更好,由于使用了注意力机制,使得9种信号预测更准确。
Vineet Thumuluri, José Juan Almagro Armenteros, Alexander Rosenberg Johansen, Henrik Nielsen, Ole Winther, DeepLoc 2.0: multi-label subcellular localization prediction using protein language models, Nucleic Acids Research, Volume 50, Issue W1, 5 July 2022, Pages W228–W234

下面开始预测,网站在这里:DeepLoc - 2.0 - Services - DTU Health Tech

从下图结果也可以看到是位于细胞核的。

排序信号重要性显示了查询蛋白中对预测具有较高重要性且与排序信号高度相关的位置的标志状图。

总结
综上所述,这三种发表在《Nucleic Acids Research》上的方法,均预测Nanog位于细胞核。
是否是糖蛋白?什么类型的糖蛋白?
GlycoEP
首先使用糖基化预测工具——GlycoEP进行预测。


从结果来看是没有N连接的,然后预测O连接同样是没有结果。
NetOGlyc、NetNGlyc
接下来使用NetOGlyc和NetNGlyc进行预测。




由上面的结果可以看到,这个蛋白是糖蛋白,N连接和O连接都存在。
亲水性和疏水性
使用ProtScale进行分析。

从下图可以知道20种氨基酸的亲疏水性,大于0为疏水,小于0为疏水。

亲水氨基酸共有153个,占比0.59。


由下图可以看到最疏水的氨基酸位于105位点,最亲水的则位于153位点。

甲虫基因
首先在ncbi上下载其蛋白质序列。

等电点?分子量?pfam保守结构域?
我们在ProtParam上进行计算,这个工具如其名,就是计算蛋白质的参数。

由计算结果可知,等电点为7.52,分子量为74351.58。
接下来使用Pfam分析蛋白质的保守结构域。


由上图可知:共有三个显著的保守结构域,分别是ABC2_membrane (PF01061)、ABC_tran (PF00005)、ABC2_membrane_7 (PF19055)。
膜蛋白质?如是则注明跨膜结构位点
使用TMHMM-2.0预测膜整合蛋白的跨膜区。
网站在这:TMHMM - 2.0 - Services - DTU Health Tech

从下面的结果可以看到,是膜蛋白质,下图的绿色框是跨膜结构位点。紫色的也是跨膜位点。

具有GPI固定(anchor)的蛋白质?
使用big-PI进行预测。
网站在这:GPI Prediction Server (imp.ac.at)

由下图可知,没有GPI固定的蛋白质。

Eisenhaber B., Bork P., Yuan Y., Loeffler G., Eisenhaber F. “Automated annotation of GPI anchor sites: case study C.elegans” TIBS (2000) 25 (7), 340-341
预测二级结构
使用Jpred进行预测。


使用PSIPRED进行预测。

由下图可以看到在序列的前半段,跨膜螺旋和Strand出现频率接近,后半段则主要是α螺旋。

对比两种方法的结果,从宏观上来看,α螺旋的分布两者是接近一致的,从微观上来看,117-125位都预测为了α螺旋,这是两个方法预测的第一个比较长的螺旋,序列后半段两者预测的也很接近。综上,可以认为两种方法预测的大体一致。
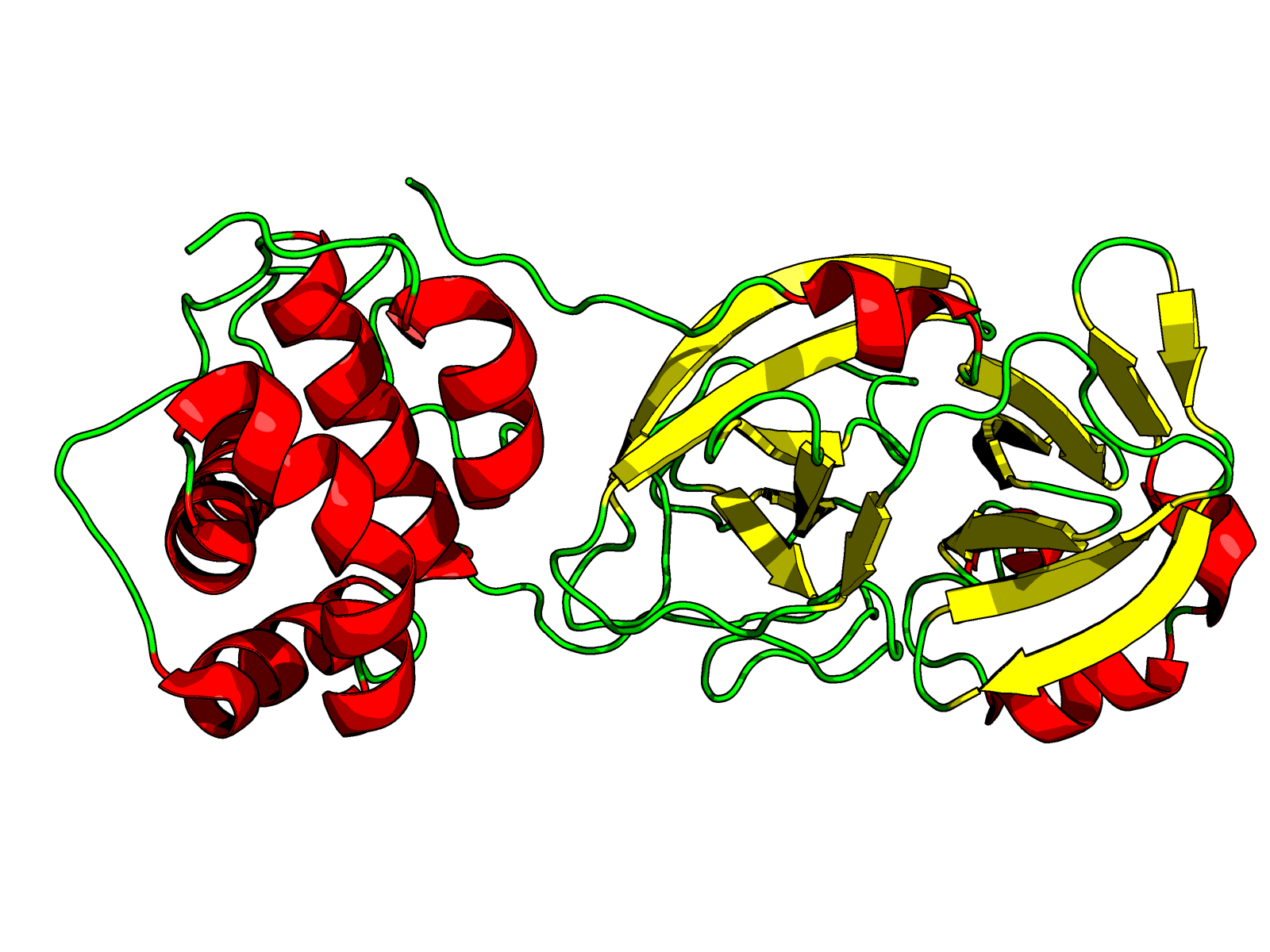
预测三级结构
使用Swiss-Model进行预测。


GMQE(全局模型质量估计)是一种结合目标-模板对齐方式和模板搜索方法的属性的质量估计。所得的GMQE分数表示为0到1之间的数字,反映了使用该对齐方式和模板构建的模型的预期准确性以及目标的覆盖范围。数字越高表示可靠性越高。可信度范围为 0-1,值越大表明质量越好。
按照GMQE从大到小进行排序,选择第一个模型。模型的指标有很多,这里最重要的是:QMEAN。QMEAN的得分可与相似大小的实验结构所期望的得分相媲美。0值附近的QMEAN 得分表明模型结构与相似大小的实验结构之间具有良好的一致性。分数为-4.0或以下表示模型的质量较低。区间-4-0,越接近0,评估待测蛋白与模板蛋白的匹配度越好。

可以看到QMEAN平均值是0.63,在350-400这个区间最小,预测的最好。

我们对拉氏图的解读,主要看模型氨基酸在几个区的分布情况。一般来说落在允许区和最大允许区的氨基酸残基占整个蛋白质的比例高于90%的,我们可以认为该模型的构象符合立体化学的规则。
坐上角是β折叠,左边中间是α螺旋,中间偏右是左旋α折叠。
由拉氏图可以知道,预测的蛋白质符合立体化学规则。

这个蛋白质在AlphaFold数据库中。网址:AlphaFold Protein Structure Database (ebi.ac.uk)

其实这个蛋白质是可以在uniProt上搜索到的,也可以在uniProt上找到其通过AlphaFold预测的三维结构

讨论
1. 结合算法原理,分析不同蛋白质二级结构预测结果的差异。
蛋白质的二级结构预测的基本依据是:每一段相邻的氨基酸残基具有形成一定二级结构的倾向,是一个模式分类问题。预测的目标是判断每一段中心的残基是否处于α螺旋、β折叠和转角(或其他状态)之一的二级结构态,即三态。
二级结构预测的方法大体分为三代:
-
统计学方法
从有限的数据集中提取各种残基形成特定二级结构的倾向,以此作为二级结构预测的依据。
-
基于单个氨基酸残基统计分析。如:Chou-Fasman法
通过统计分析,获得每个残基出现于特定二级结构构象的倾向性因子,进而利用这些倾向性因子预测蛋白质的二级结构。
-
基于氨基酸片段的统计分析。如:GOR方法
GOR是一种基于信息论和贝叶斯统计学的方法,将蛋白质序列当作一连串的信息值来处理。不仅考虑被预测位置本身氨基酸种类的影响,而且考虑相邻残基种类对该位置构象的影响,准确率大约为65%。
-
-
基于立体化学原则的物理化学方法
比如Lim方法。因为氨基酸的理化性质对二级结构影响较大,在进行结构预测时要考虑氨基酸残基的物理化学性质。比如疏水性、极性、侧链基团的大小等。
-
神经网络与人工智能
结构预测还有同源分析法,也就是将待预测的片段与数据库中已知二级结构的片段进行相似性比较,利用打分矩阵计算出相似性得分。如果数据库中有相似性大于30%的序列,则预测准确率可以大大上升。
可以对同源分析之后的结果构建一个神经网络模型进行预测。
在本次实验中,我们用了JPred和PSIPRED方法进行预测。
JPred4使用了Jnet算法,是一个输入多序列比对的结果(可用PSIBLAST和HMM)到神经网络中的预测方法。

Drozdetskiy A, Cole C, Procter J, Barton GJ. JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 2015 Jul 1;43(W1):W389-94. doi: 10.1093/nar/gkv332. Epub 2015 Apr 16. PMID: 25883141; PMCID: PMC4489285.
PSIPRED(PSI-blast based secondary structure PREDiction )也是一个利用神经网络的方法。

这两种方法用的数据和方法比较相近,从实验的结果来看也比较相近。
2. 蛋白质的类型、序列长度等哪些自身因素有可能影响蛋白质结构预测的效果?
- 蛋白质类型:不同类型的蛋白质可能具有不同的结构特征,因此对于不同类型的蛋白质,结构预测的效果也可能不同。例如,对于膜蛋白来说,它们的结构预测效果可能会比普通的水溶性蛋白要差一些。
- 蛋白质序列长度:蛋白质的序列长度也可能会影响结构预测的效果。通常来说,蛋白质序列越长,结构预测的效果就越差。这是因为蛋白质序列越长,就意味着它可能具有更复杂的结构,从而导致结构预测的难度更大。
- 其他自身因素:疏水性、氢键、二硫键的形成、静电作用、范德华力以及溶剂作用。可以针对范德华力、氢键、溶剂、静电和其它力对一个折叠蛋白总体稳定性的相对作用来建立能量函数,找到能量函数的最低状态。
3. 为什么需要对蛋白质的亚细胞定位进行预测?
亚细胞定位是指某种蛋白或某个基因表达产物在细胞内的具体存在部位,如在胞核,胞浆内,细胞膜或某一特定细胞器上存在。蛋白分布在不同细胞的不同部位,对蛋白的亚细胞定位分析有助于蛋白功能研究的初步判断,同时为理解基因的作用机制提供研究方向。简而言之就是有助于了解蛋白质功能与互作。
4. 通过动手实践分析,你对哪个分析背后的基本原理有了更深入的理解?
WoLF PSORT用了之前PSORT的一些特征,以及新加进去的氨基酸组成的特征,然后利用加权的KNN进行分类。使用了最相关的特征,使得模型不容易过拟合,同时也使得模型更容易解释。
预测的难度更大。
3. 其他自身因素:疏水性、氢键、二硫键的形成、静电作用、范德华力以及溶剂作用。可以针对范德华力、氢键、溶剂、静电和其它力对一个折叠蛋白总体稳定性的相对作用来建立能量函数,找到能量函数的最低状态。
3. 为什么需要对蛋白质的亚细胞定位进行预测?
亚细胞定位是指某种蛋白或某个基因表达产物在细胞内的具体存在部位,如在胞核,胞浆内,细胞膜或某一特定细胞器上存在。蛋白分布在不同细胞的不同部位,对蛋白的亚细胞定位分析有助于蛋白功能研究的初步判断,同时为理解基因的作用机制提供研究方向。简而言之就是有助于了解蛋白质功能与互作。
4. 通过动手实践分析,你对哪个分析背后的基本原理有了更深入的理解?
WoLF PSORT用了之前PSORT的一些特征,以及新加进去的氨基酸组成的特征,然后利用加权的KNN进行分类。使用了最相关的特征,使得模型不容易过拟合,同时也使得模型更容易解释。

开放原子开发者工作坊旨在鼓励更多人参与开源活动,与志同道合的开发者们相互交流开发经验、分享开发心得、获取前沿技术趋势。工作坊有多种形式的开发者活动,如meetup、训练营等,主打技术交流,干货满满,真诚地邀请各位开发者共同参与!
更多推荐
 9
9 0
0- 0



 已为社区贡献10条内容
已为社区贡献10条内容

所有评论(0)