一文解决Cellphonedb单细胞互作分析及可视化作图(2)
我们在运行完cellphonedb的热图作图函数后,热图展示的已经是筛选过的p
之前我们已经讲述了Cellphonedb的安装配置和数据分析,就差可视化了,这里简单说一下常见的可视化,如果需要其他更加个性化的可视方式,就需要发挥自己的聪明才智了!
CellphoneDB单细胞互作分析:
CellPhoneDB单细胞互作分析(1):Linux软件安装及遇到的Bug(ERROR)解决
CellPhoneDB单细胞互作分析(2):数据分析|人鼠基因同源转化|ERROR解决|详细注释版代码
cellphonedb分析完成后,我们得到6个数据文件,将其全部读入,看似6个,从内容上讲其实就三个。利用这些文件就可以做互作网络图、受配体气泡图及热图等等文章中常见的可视化图形了。
setwd("D:/cellinter-celldb/out")
#cellphonedb可视化
count_net <- read.delim("count_network.txt", check.names = FALSE)
inter_net <- read.delim("interaction_count.txt", check.names = FALSE)
pvalues <- read.delim("pvalues.txt", check.names = FALSE)
means <- read.delim("means.txt", check.names = FALSE)
sig.means <- read.delim("significant_means.txt", check.names = FALSE)
deconvoluted <- read.delim("deconvoluted.txt", check.names = FALSE)
一、互作网络图
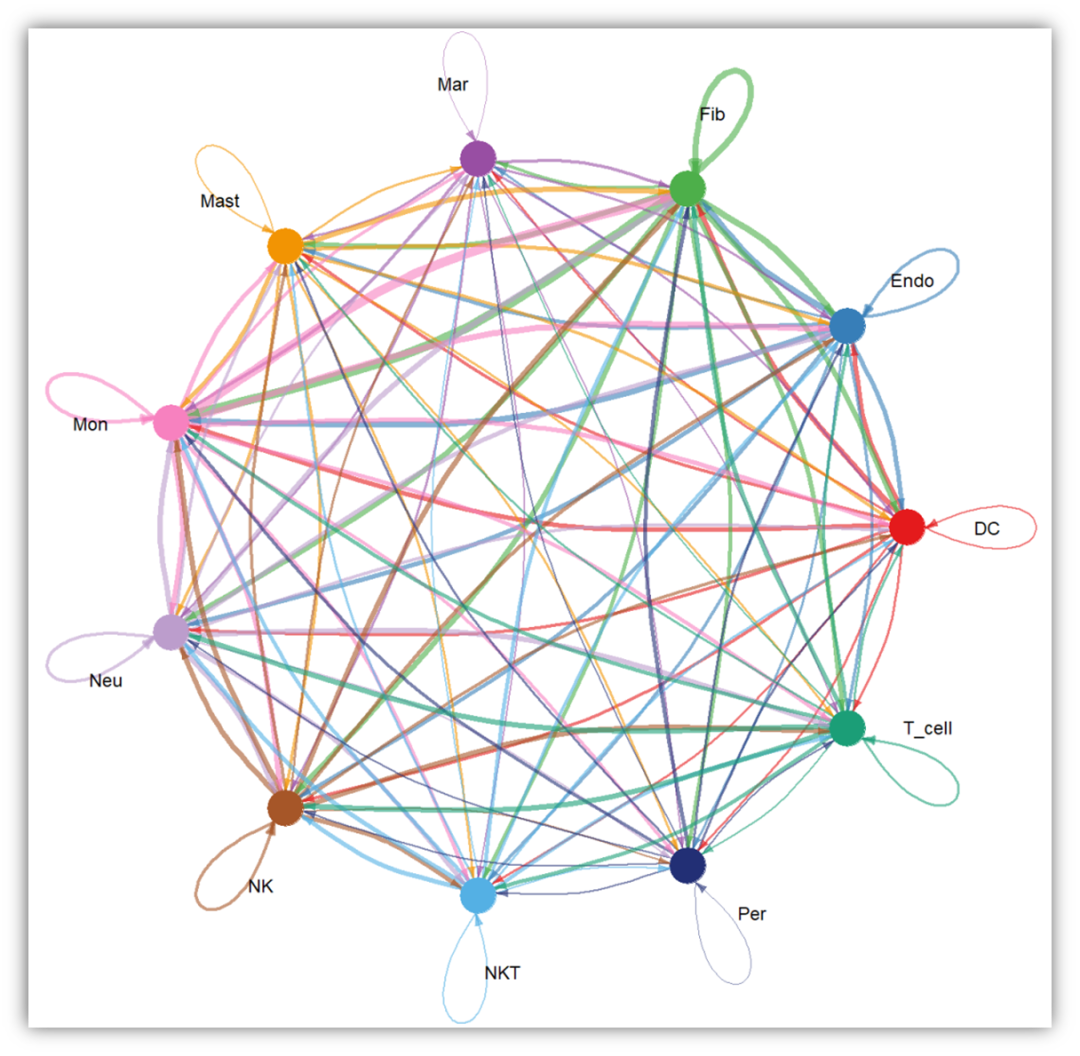
互作图很多网上教程使用igraph包做的,其实这是源码,确实是需要这样做的,但是我认为太麻烦,因为你要自己调整,还不一定做的对。所以我想了一个妙招---”借鸡下蛋“,这应该是生信人需要做的举一反三。之前我们说过Cellchat的单细胞互作分析(Cellchat(代码详细注释版):单细胞转录组(人、小鼠)细胞互作分析及可视化),这里面有作者写好的函数,可视化也是相当不错的,只要将Cellphonedb文件格式整理成需要的格式,使用Cellchat函数包不就完成了?需要的文件是count_network.txt
#互作网络图
library(CellChat)
count_inter <- count_net
count_inter$count <- count_inter$count/100
library(tidyr)
count_inter<-spread(count_inter, TARGET, count)
rownames(count_inter) <- count_inter$SOURCE
count_inter <- count_inter[, -1]
count_inter <- as.matrix(count_inter)
netVisual_circle(count_inter,weight.scale = T)

image.png
每种细胞与其他细胞的互作,循环出图!
par(mfrow = c(4,3), xpd=TRUE)
for (i in 1:nrow(count_inter)) {
mat2 <- matrix(0, nrow = nrow(count_inter), ncol = ncol(count_inter), dimnames = dimnames(count_inter))
mat2[i, ] <- count_inter[i, ]
netVisual_circle(mat2,
weight.scale = T,
edge.weight.max = max(count_inter),
title.name = rownames(count_inter)[i],
arrow.size=0.2)
}

image.png
二、热图
其实热图和网络图一样,都是展示了不同细胞之间互作的数目。我们在运行完cellphonedb的热图作图函数后,热图展示的已经是筛选过的p<0.05的受配体互作数目,生成的文件是count_net, 这里我们直接用这个文件作热图,进行一些简单的修饰,例如展示互作数目,其他个性化修饰同热图。当然如果不需要默认的作图,自己筛选做热图需要从pvalues这个文件开始,具体可参考网上其他教程。
#热图其实和网络图一样,都是展示互作数目的。
library(tidyr)
count_matrix<-spread(count_net, TARGET, count)
rownames(count_matrix) <- count_matrix$SOURCE
count_matrix <- count_matrix[, -1]
count_matrix <- as.matrix(count_matrix)
用pheatmap做热图。
library(pheatmap)
pheatmap(count_matrix, show_rownames = T, show_colnames = T, scale="none", cluster_cols = T,
border_color='white', cluster_rows = T, fontsize_row = 14, fontsize_col = 14,
main = "Control", treeheight_row = 0, family = 'Arial',
color = colorRampPalette(c("dodgerblue4",'peachpuff','deeppink4' ))( 1000 ),
treeheight_col = 0,
display_numbers = T, number_color="white",fontsize_number=12,
number_format="%.0f", legend_labels = c(0,300))

image.png
三、受配体点图
受配体点图的绘制需要两个文件pvalues.txt和means.txt,原理是将两个文件合并,提取受配体对进行可视化,ggplot就可以实现。但是自己弄比较繁琐,这里有一个专门针对cellphonedb的可视化R包,可以轻松解决这一问题,你只需修改参数即可,可视化效果也不错,用在文章中绰绰有余!
#安装
if (!requireNamespace("devtools", quietly = TRUE))
install.packages("devtools")
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
devtools::install_github('zktuong/ktplots', dependencies = TRUE)
library(ktplots)
#作图需要的文件,means.txt, pvalues.txt, 单细胞seurat对象
读入作图需要的文件。
pvals <- read.delim(paste0(pbmc,"pvalues.txt"), check.names = FALSE)
means <- read.delim(paste0(pbmc,"means.txt"), check.names = FALSE)
load("E:/生物信息学/数据/scRNA.Rdata")
气泡图。cell_type1、2指定互作细胞,如果不指定则默认所有细胞,这里我们指定DC,可视化的就是DC发出信号作用于其他细胞,以及DC接受的其他细胞的作用。实际情况按照自己的实验目的指定。scdata就是我们用于分析的单细胞seurat对象。means和pvals就是我们cellphonedb分析产生的两个文件。gene.family是一些基因对集合,有chemokines', 'Th1', 'Th2', 'Th17', 'Treg', 'costimulatory', 'coinhibitory', 'niche',可以选择需要的指定,不选择则默认所有。keep_significant_only=T,我们只选择显著的。其他的可视化参数可自行查阅函数,可视化是基于ggplot的,所以主题修改和ggplot一样。
plot_cpdb(cell_type1 = 'DC', cell_type2 = "", scdata = scRNA,
idents = 'celltype', means = means, pvals = pvals,
gene.family = 'costimulatory',highlight = "blue",
keep_significant_only=T) +
theme(axis.text = element_text(size = 10, color = 'black'))

image.png
这样的可视化就很足够了。当然了,ktplots还有其他的可视化,比如比较新奇的将细胞之间互作基因对用弦图展示,还可以指定特定的几种细胞互作弦图展示。还可以多组分析可视化、结合空间转录组的可视化、对于单细胞的可能化等等,感兴趣的值得学习。但是就我们细胞互作而言,已经足够了。
我们这里提供了最少的代码和步骤,比较简洁的完成了cellphonedb的可视化,我觉得这就足够了。如果你觉得有用,分享点赞一下呗!当然,你可以学习其他包的函数,研究明白其原理,可以自己写函数可视化。
更多精彩内容请至我的公众号《KS科研分享与服务》

开放原子开发者工作坊旨在鼓励更多人参与开源活动,与志同道合的开发者们相互交流开发经验、分享开发心得、获取前沿技术趋势。工作坊有多种形式的开发者活动,如meetup、训练营等,主打技术交流,干货满满,真诚地邀请各位开发者共同参与!
更多推荐


 9
9 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)