
计算物理模块pymatgen的安装配置,使用教程与示例分析(全网最全?)
最近在学pymatgen,但是发现网上根本没多少正经的教程,要么机翻,要么chatgpt,我总结了一下部分比较好的教程,写成了这篇教程,可能是全网最全?至少我是这么认为的
最近在学pymatgen,但是发现网上根本没多少正经的中文教程,要么机翻,要么chatgpt,我总结了一下部分比较好的教程,写成了这篇中文教程,可能是全网最全?至少在内网是这样的
但是本文只能用于新手入门,如果你需要实际的计算工作还是需要自己再摸索一下XD
安装与配置
首先安装python,可以直接前往python官网下载安装包后安装,建议3.8.X,这里并没有很多需要注意的,只需要注意在安装的时候不要忘记将其加入系统变量,即框选以下的框框,不然后面还要手动配置系统环境变量会很烦

安装完成后如果没有其他需求可以直接点击win+r输入cmd进入命令台
在命令台中输入下述指令完成pymatgen的下载
pip install pymatgen但是如果你本身就已经在使用Python做一些项目了,我建议你使用虚拟环境,即使用conda,这边推荐使用miniforge3,也可以自己百度搜到然后下载,也是别忘了加入系统变量,然后同样的进入命令台
conda create -n 你想要的环境名称 python=3.8 #创建虚拟环境
conda activate 刚才设置的环境名称 #进入虚拟环境
pip install 刚才设置的环境名称 #安装pymatgen此时就已经安装完成了最基本的pymatgen,但是如果想要进行更加全面的功能使用,如VASP运算,就需要进行下一步编译工作()
基本模块介绍
1. 核心模块(Core Modules)
-
Structure: 用于表示和操作晶体结构。它支持读取和写入多种格式的结构文件(如CIF、POSCAR等),并提供了一系列方法来分析和处理晶体结构,如计算晶胞参数、体积、形状、空间群等。
-
Element/Species: 用于表示化学元素和物种(如离子、分子等)。这些类提供了元素的基本属性(如原子质量、电负性等)和物种的复合属性(如化合价、电荷等)。
2. 电子结构分析模块(Electronic Structure Analysis Modules)
-
BandStructure: 用于处理和分析能带结构数据。它支持绘制能带图和态密度图,并提供了计算能带宽度、有效质量等性质的工具。
-
DOS: 用于处理和分析态密度(Density of States, DOS)数据。它可以与BandStructure模块结合使用,以更全面地分析材料的电子结构。
3. 材料性质预测模块(Material Properties Prediction Modules)
-
MPRester: 集成了Materials Project REST API,允许用户通过pymatgen直接访问Materials Project的在线数据库,以获取材料的电子和热力学性质,如电荷密度分布、费米面、热膨胀系数、熔点等。
-
Thermo: 提供了计算材料热力学性质的工具,如熵、焓、吉布斯自由能等。
4. 高通量计算模块(High-throughput Computation Modules)
-
Mapper: 提供了高通量计算的框架,用于自动执行各种计算任务(如DFT计算)并管理大量计算结果。它支持多种计算方法和代码库(如VASP、QE等),并允许用户通过脚本或命令行界面轻松地设置和运行计算任务。
-
Machine Learning: 集成了机器学习算法,用于对大量计算结果进行挖掘和分类,以加速材料筛选和优化过程。
5. 结构和晶体学分析模块(Structure and Crystallography Analysis Modules)
-
Symmetry: 用于分析晶体结构的对称性,包括确定空间群、对称操作等。它支持多种对称性分析方法,并提供了生成对称元素和对称操作列表的工具。
-
Diffraction: 提供了计算晶体衍射图谱的工具,如布喇格散射图等。这些工具可以帮助用户理解和分析晶体结构的衍射行为。
6. 转换和生成模块(Transformation and Generation Modules)
-
Transformations: 提供了一系列用于结构转换的方法(类),如掺杂、建超胞、添加氧化态等。这些类可以轻松地实现结构的修改和优化。
-
Generators: 提供了生成特定类型结构(如表面、界面、缺陷等)的工具。这些工具可以帮助用户构建复杂的材料模型以进行进一步的研究和分析。
7. 可视化模块(Visualization Modules)
-
Plotting: pymatgen支持通过matplotlib等库绘制高质量的晶体结构图、能带图、态密度图等。这些可视化工具可以帮助用户更直观地理解材料结构和性质之间的关系。
基本用法
参考网站,请各位积极支持原作者
化学物质基本信息输出
####引用pymatgen####
import pymatgen.core as mg
####设置研究对象为硅Si####
si = mg.Element("Si")
####输出硅的原子质量####
print(f"Atomic mass of Si is {si.atomic_mass}")
####输出硅的熔点####
print(f"Si has a melting point of {si.melting_point}")
####输出离子硅的半径####
print(f"Ionic radii for Si: {si.ionic_radii}")运行后即可输出
Atomic mass of Si is 28.0855 amu
Si has a melting point of 1687.0 K
Ionic radii for Si: {4: 0.54}其中第一行为硅的原子质量,第二行为硅的熔点,第三行为离子硅的半径,即+4价硅离子的半径为0.54 Å。
如果你想要其他单位的原子质量可以将质量代码改为以下内容(即改为kg单位)
print("Atomic mass of Si in kg: {}".format(si.atomic_mass.to("kg")))
###输出结果###
Atomic mass of Si in kg: 4.66370699549943e-26 kg如果你想分别输出价态和离子半径可以把上面的半径代码改为下述内容,即检阅离子半径内容并将其分为价态与半径两部分,可以进行分别输出。
s1=si.ionic_radii
for charge, radius in s1.items():
print(f"Si^{charge} 的离子半径为 {radius} Å")
###输出结果###
Si^4 的离子半径为 0.54 ang Å进一步的,也可以直接输出离子的信息,如下述内容(二价铁离子)
##设置2价铁离子为研究对象##
fe2 = mg.Species("Fe", 2)
##输出铁离子的离子质量##
print(fe2.atomic_mass)
##输出铁离子的离子半径##
print(fe2.ionic_radius)
##输出内容##
55.845 amu
0.92 ang当然我们的研究对象也不能只是单质,也可以是化合物等,我们以二氧化三铁为例
##引用pymatgen##
import pymatgen.core as mg
##设置二氧化三铁为研究对象##
comp = mg.Composition("Fe2O3")
##输出二氧化三铁的质量##
print(f"Weight of Fe2O3 is {comp.weight}")
##输出二氧化三铁中铁的个数##
print("Amount of Fe in Fe2O3 is {}".format(comp["Fe"]))
##输出二氧化三铁中铁的原子分数##
print("Atomic fraction of Fe is {}".format(comp.get_atomic_fraction("Fe")))
##输出二氧化三铁中铁的重量分数##
print("Weight fraction of Fe is {}".format(comp.get_wt_fraction("Fe")))
##输出内容##
Weight of Fe2O3 is 159.6882 amu
Amount of Fe in Fe2O3 is 2.0
Atomic fraction of Fe is 0.4
Weight fraction of Fe is 0.699425505453753 物质结构信息
在材料科学中,晶格(Lattice)代表布拉维晶格(Bravais lattice),它是一种理想的、无限重复的点阵结构。pymatgen库提供了方便的静态函数来创建常见的晶格类型。
接下来我们使用pymatgen创建一个立方晶格,其中cubic为立方晶格,4,2为立方晶格的晶格常数
# 引用pymatgen #
from pymatgen.core import Lattice, Structure, IStructure
# 创建一个具有晶格参数4.2的立方晶格
lattice = Lattice.cubic(4.2)
print(lattice.parameters) # 打印晶格参数
#输出内容#
(4.2, 4.2, 4.2, 90.0, 90.0, 90.0)如果不想要既定的晶格,想要更加方便的自定义晶格,可以直接使用矩阵进行晶格设置,如立方晶格
from pymatgen.core import Lattice
# 创建立方晶格的矩阵
lattice_matrix = [
[4.2, 0, 0],
[0, 4.2, 0],
[0, 0, 4.2]
]
# 创建Lattice对象
lattice = Lattice(lattice_matrix)
# 打印晶格参数
print(lattice)
#输出内容#
4.200000 0.000000 0.000000
0.000000 4.200000 0.000000
0.000000 0.000000 4.200000或三斜晶格
from pymatgen.core import Lattice
# 创建三斜晶格的矩阵
lattice_matrix = [
[5.0, 0, 0],
[2.0, 4.0, 0],
[1.0, 1.0, 5.0]
]
# 创建Lattice对象
lattice = Lattice(lattice_matrix)
# 打印晶格参数
print(lattice)
print("alpha =", lattice.alpha)
print("beta =", lattice.beta)
print("gamma =", lattice.gamma)
#输出内容#
5.000000 0.000000 0.000000
2.000000 4.000000 0.000000
1.000000 1.000000 5.000000
alpha = 75.03678256669288
beta = 78.90419671686361
gamma = 63.43494882292201
接下来我们创建一个包含铯(Cs)和氯(Cl)原子的晶体结构
from pymatgen.core import Lattice, Structure
import pymatgen.core as mg
#创建一个立方晶格#
lattice = mg.Lattice.cubic(4.2)
# 创建一个包含 Cs 和 Cl 原子的晶体结构
# 原子位于 (0, 0, 0) 和 (0.5, 0.5, 0.5) 分数坐标
structure = Structure(lattice, ["Cs", "Cl"], [[0, 0, 0], [0.5, 0.5, 0.5]])
# 打印单元格体积
print(f"Unit cell vol = {structure.volume}")
# 打印结构中第一个原子的信息
print(f"First site of the structure is {structure[0]}")
#输出内容#
Unit cell vol = 74.08800000000001
First site of the structure is [0. 0. 0.] Cs不仅如此,Structure 对象包含了许多用于操作晶体结构的有用函数。这些操作可以改变结构中的原子种类、位置和数量,如:
make_supercell:通过给定的倍数扩展原始晶胞,创建一个超胞。del structure[0]:删除结构中的第一个原子。append:向结构中添加新的原子。structure[-1] = "Li":更改最近添加的原子种类。structure[0] = "Cs", [0.01, 0.5, 0]:移动第一个原子的位置。IStructure.from_sites(structure):创建一个不可变的晶体结构对象。
# 创建结构的 3 x 2 x 1 超胞
structure.make_supercell([2, 2, 1])
# 删除第一个位点
del structure[0]
# 追加一个 Na 原子
structure.append("Na", [0, 0, 0])
# 将最后添加的原子更改为 Li
structure[-1] = "Li"
# 将第一个原子在 x 方向上移动 0.01 的分数坐标
structure[0] = "Cs", [0.01, 0.5, 0]
# 创建一个不可变的晶体结构(不能修改)
immutable_structure = mg.IStructure.from_sites(structure)
# 打印不可变结构的信息
print(immutable_structure)将该代码放在我们前文的CsCl代码后可以得到以下结果
Full Formula (Cs3 Li1 Cl4)
Reduced Formula: Cs3LiCl4
abc : 8.400000 8.400000 4.200000
angles: 90.000000 90.000000 90.000000
pbc : True True True
Sites (8)
# SP a b c
--- ---- ---- ---- ---
0 Cs 0.01 0.5 0
1 Cs 0.5 0 0
2 Cs 0.5 0.5 0
3 Cl 0.25 0.25 0.5
4 Cl 0.25 0.75 0.5
5 Cl 0.75 0.25 0.5
6 Cl 0.75 0.75 0.5
7 Li 0 0 0这段代码演示了如何对晶体结构进行一系列修改,包括创建超胞、删除和添加原子以及修改原子位置。然后在最后创建了一个不可变的晶体结构对象,该对象一旦创建就不能被更改。这种不可变对象在需要确保结构数据不被意外修改的情况下非常有用。
不仅如此,pymatgen还能给出其他基本信息数据,下面以NaCl为例
from pymatgen.core.structure import Structure
# 定义晶体结构
nacl_structure = Structure(
lattice=[[5.64, 0, 0], [0, 5.64, 0], [0, 0, 5.64]],
species=["Na", "Cl"],
coords=[[0, 0, 0], [0.5, 0.5, 0.5]])
# 打印晶体结构
print(nacl_structure)
# 获取晶体结构的元素种类
print(nacl_structure.symbol_set)
# 获取晶体结构的元素种类及数量
print(nacl_structure.composition.items())
# 获取晶体结构的分数坐标
print(nacl_structure.frac_coords)
# 获取晶体结构的笛卡尔坐标
print(nacl_structure.cart_coords)
# 获取晶体结构的晶胞
print(nacl_structure.lattice)
# 获取晶体结构的体积
print(nacl_structure.volume)
# 获取晶体结构的密度
print(nacl_structure.density)
# 获取晶体结构的平均分子体积
print(nacl_structure.volume / nacl_structure.num_sites)
# 获取晶体结构的平均分子密度
print(nacl_structure.density / nacl_structure.composition.num_atoms)
#获取晶体结构的空间群信息
print(nacl_structure.get_space_group_info())基本分析
pymatgen 提供了许多用于分析晶体结构的函数。如
from pymatgen.symmetry.analyzer import SpacegroupAnalyzer
finder = SpacegroupAnalyzer(structure)
print(f"The spacegroup is {finder.get_space_group_symbol()}")
其中的SpacegroupAnalyzer 可以用来确定晶体结构的空间群,当把上述代码放在我们前文CsCl后,可以得到结果
The spacegroup is Pmm2from pymatgen.analysis.structure_matcher import StructureMatcher
# 创建两个拓扑相同但元素不同且一个晶格更大的结构
s1 = mg.Structure(lattice, ["Cs", "Cl"], [[0, 0, 0], [0.5, 0.5, 0.5]])
s2 = mg.Structure(mg.Lattice.cubic(5), ["Rb", "F"], [[0, 0, 0], [0.5, 0.5, 0.5]])
# 初始化结构匹配器
m = StructureMatcher()
# 进行结构匹配
print(m.fit_anonymous(s1, s2)) # 返回一个映射,将 s1 和 s2 对应起来。也可以使用严格的元素匹配。其中的StructureMatcher,它可以比较两个结构是否拓扑相同,即使它们的元素类型或晶格参数不同,运行以上代码,可以得到以下结果
True数据输入与结果输出
如pymatgen 提供了对多种文件格式的输入输出支持,这些功能集中在 pymatgen.io 包中。
# 将结构保存为不同格式的文件
structure.to(filename="POSCAR")
structure.to(filename="CsCl.cif")也可以同前文展示一样直接打印出结果
print(structure.to(fmt="poscar"))
print(structure.to(fmt="cif"))可以直接读取文件中的数据结构
from pymatgen import Structure
# 从文件中读取结构
structure = Structure.from_file("POSCAR")也可以生成VASP文件,vaspio_set 模块提供了一种获取完整VASP输入文件集的方法,以便进行计算。这段代码将为给定的结构生成一套完整的VASP输入文件,并将它们写入名为 MyInputFiles 的目录中。
from pymatgen.io.vasp.sets import MPRelaxSet
# 创建一个 MPRelaxSet 对象
v = MPRelaxSet(structure)
# 将完整的输入文件集写入指定目录
v.write_input("MyInputFiles")我们以NaCl为例,以下为输出文件代码
from pymatgen.core.structure import Structure
# 定义晶体结构
nacl_structure = Structure(
lattice=[[5.64, 0, 0], [0, 5.64, 0], [0, 0, 5.64]],
species=["Na", "Cl"],
coords=[[0, 0, 0], [0.5, 0.5, 0.5]])
# 将晶体结构保存为POSCAR文件
nacl_structure.to(fmt="poscar", filename="POSCAR")
# 将晶体结构保存为CIF文件
nacl_structure.to(fmt="cif", filename="NaCl.cif")然后我们也可以读取输出的文件
from pymatgen.core import Structure
from pymatgen.symmetry.analyzer import SpacegroupAnalyzer
#读取刚才输出的cif文件#
structure= Structure.from_file('NaCl.cif')
#获得相关信息#
primitive_structure = structure.get_primitive_structure()
# 获取空间群名称
space_group_name = sg_analyzer.get_space_group_symbol()
# 获取空间群编号
space_group_number = sg_analyzer.get_space_group_number()
#打印#
print(primitive_structure)
print("空间群名称:", space_group_name)
print("空间群编号:", space_group_number)运行后可以得到我们写入的NaCl相关信息
Full Formula (Na1 Cl1)
Reduced Formula: NaCl
abc : 5.640000 5.640000 5.640000
angles: 90.000000 90.000000 90.000000
pbc : True True True
Sites (2)
# SP a b c
--- ---- --- --- ---
0 Na 0 0 0
1 Cl 0.5 0.5 0.5
空间群名称: Pm-3m
空间群编号: 221画图
此外,由于pymatgen自带matplotlib,所以可以进行画图功能,但是有些时候会由于版本问题无法进行绘制,以下是示例,用于生成金(Au)晶体的表面模型,并使用 pymatgen 的 AdsorbateSiteFinder 来寻找可能的吸附位点
from pymatgen.core.surface import generate_all_slabs
from pymatgen.analysis.adsorption import *
import matplotlib.pyplot as plt
from pymatgen.core.structure import Structure
# 创建金晶体结构
structure = Structure(
lattice=[[4.17, 0, 0], [0, 4.17, 0], [0, 0, 4.17]],
species=["Au"]*4,
coords=[[0.0, 0.0, 0.0],
[0.5, 0.5, 0.0],
[0.5, 0.0, 0.5],
[0.0, 0.5, 0.5]])
print(structure)
# 生成表面模型
slabs = generate_all_slabs(structure, max_index=1, min_slab_size=8.0, min_vacuum_size=15.0)
# 筛选出 (1,0,0) 晶面的表面模型
Ag_100= [slab for slab in slabs if slab.miller_index==(1,0,0)][0]
# 寻找吸附位点
asf_Ag_100 = AdsorbateSiteFinder(Ag_100)
ads_sites = asf_Ag_100.find_adsorption_sites()
print(ads_sites)
# 绘制表面模型和吸附位点
fig = plt.figure(facecolor='gray', edgecolor='red')
ax = fig.add_subplot(111)
plot_slab(Ag_100, ax, adsorption_sites=True)
plt.show()输出结果为
示例
参考网站,请各位积极支持原作者Pymatgen的学习以及应用 - PhoenixJasonPymatgen的学习以及应用 Pymatgen对于材料计算来说是一个非常有用的库,我前段时间基于这个库写了一个有关层状氧化物生成POSCAR的脚本,具体来说是使用了里面对Ewald能量的求解来帮我初步筛选一些能量,不过这个库的确是可以非常便捷的帮我编辑结构。 由于对这个库的使用一部分上涉及到了我课题的核心部分,所以代码肯定是不能全部放出来了,但是我会放出部分代码来作为教学,记录一下,此外也会![]() https://phoenixjason.cn/2023/05/31/20230531Pymatgen%E7%9A%84%E5%AD%A6%E4%B9%A0%E4%BB%A5%E5%8F%8A%E5%BA%94%E7%94%A8/作者在这篇文章中主要就先搭建一个模型,然后用 pymategn读取这个数据,在里面随机插入Na离子后进行Eward能量计算并保存数据
https://phoenixjason.cn/2023/05/31/20230531Pymatgen%E7%9A%84%E5%AD%A6%E4%B9%A0%E4%BB%A5%E5%8F%8A%E5%BA%94%E7%94%A8/作者在这篇文章中主要就先搭建一个模型,然后用 pymategn读取这个数据,在里面随机插入Na离子后进行Eward能量计算并保存数据
所以主要内容分为,读取,随机生成并插入,计算,保存。其中读取和保存我们已经给过实现方法了。
首先以下为原文作者给出的模型,其中模型名称为POSCAR

test.xsd
1.00000000000000
7.4669587686781895 -4.3110504950760768 -0.0000000000000000
0.0000000000000000 8.6221009901521537 0.0000000000000000
0.0000000000000000 -0.0000000000000000 9.7568998343236562
O Mn
36 18
Direct
0.1111110000000011 0.2222220000000021 0.0992854113435739
0.2222220000000021 0.1111110000000011 0.5992854113435734
0.2222220000000021 0.1111110000000011 0.9007145886564266
0.1111110000000011 0.2222220000000021 0.4007145886564263
0.4444439999999972 0.2222220000000021 0.0992854113435739
0.5555560000000028 0.1111110000000011 0.5992854113435734
0.5555560000000028 0.1111110000000011 0.9007145886564266
0.4444439999999972 0.2222220000000021 0.4007145886564263
0.7777779999999979 0.2222220000000021 0.0992854113435739
0.8888889999999989 0.1111110000000011 0.5992854113435734
0.8888889999999989 0.1111110000000011 0.9007145886564266
0.7777779999999979 0.2222220000000021 0.4007145886564263
0.1111110000000011 0.5555560000000028 0.0992854113435739
0.2222220000000021 0.4444439999999972 0.5992854113435734
0.2222220000000021 0.4444439999999972 0.9007145886564266
0.1111110000000011 0.5555560000000028 0.4007145886564263
0.4444439999999972 0.5555560000000028 0.0992854113435739
0.5555560000000028 0.4444439999999972 0.5992854113435734
0.5555560000000028 0.4444439999999972 0.9007145886564266
0.4444439999999972 0.5555560000000028 0.4007145886564263
0.7777779999999979 0.5555560000000028 0.0992854113435739
0.8888889999999989 0.4444439999999972 0.5992854113435734
0.8888889999999989 0.4444439999999972 0.9007145886564266
0.7777779999999979 0.5555560000000028 0.4007145886564263
0.1111110000000011 0.8888889999999989 0.0992854113435739
0.2222220000000021 0.7777779999999979 0.5992854113435734
0.2222220000000021 0.7777779999999979 0.9007145886564266
0.1111110000000011 0.8888889999999989 0.4007145886564263
0.4444439999999972 0.8888889999999989 0.0992854113435739
0.5555560000000028 0.7777779999999979 0.5992854113435734
0.5555560000000028 0.7777779999999979 0.9007145886564266
0.4444439999999972 0.8888889999999989 0.4007145886564263
0.7777779999999979 0.8888889999999989 0.0992854113435739
0.8888889999999989 0.7777779999999979 0.5992854113435734
0.8888889999999989 0.7777779999999979 0.9007145886564266
0.7777779999999979 0.8888889999999989 0.4007145886564263
0.0000000000000000 0.0000000000000000 -0.0000000000000000
0.0000000000000000 0.0000000000000000 0.5000000000000000
0.3333330000000032 0.0000000000000000 -0.0000000000000000
0.3333330000000032 0.0000000000000000 0.5000000000000000
0.6666669999999968 0.0000000000000000 -0.0000000000000000
0.6666669999999968 0.0000000000000000 0.5000000000000000
0.0000000000000000 0.3333330000000032 -0.0000000000000000
0.0000000000000000 0.3333330000000032 0.5000000000000000
0.3333330000000032 0.3333330000000032 -0.0000000000000000
0.3333330000000032 0.3333330000000032 0.5000000000000000
0.6666669999999968 0.3333330000000032 -0.0000000000000000
0.6666669999999968 0.3333330000000032 0.5000000000000000
0.0000000000000000 0.6666669999999968 -0.0000000000000000
0.0000000000000000 0.6666669999999968 0.5000000000000000
0.3333330000000032 0.6666669999999968 -0.0000000000000000
0.3333330000000032 0.6666669999999968 0.5000000000000000
0.6666669999999968 0.6666669999999968 -0.0000000000000000
0.6666669999999968 0.6666669999999968 0.5000000000000000随后便为读取内容
from pymatgen.core.structure import Structure
from pymatgen.io.vasp import Poscar
def get_site(structure, element):
# 获取结构中Mn的Site信息
mn_sites = [site for site in structure if site.specie.symbol == element]
# 获取结构中Mn的坐标信息
mn_coords = [site.frac_coords for site in mn_sites]
# 获取结构中Mn的z轴坐标
mn_z_coords = [site.frac_coords[2] for site in mn_sites]
# 对结构中Mn的z轴坐标进行归一处理
mn_z_coords = [1 + i if i <= 0.25 else i for i in mn_z_coords]
# 对结构中Mn原子进行分层处理
mn_layer1 = [i for i in mn_z_coords if i > 0.75]
mn_layer2 = [i for i in mn_z_coords if i <= 0.75]
# 按照层将每层Mn原子的z轴坐标提取出来
mn_layer1_site = [mn_coords[i] for i, z_coord in enumerate(mn_z_coords) if z_coord > 0.75]
mn_layer2_site = [mn_coords[i] for i, z_coord in enumerate(mn_z_coords) if z_coord <= 0.75]
# 求解Mn层的平均z轴坐标
layer1_avr_z = sum(mn_layer1) / len(mn_layer1)
layer2_avr_z = sum(mn_layer2) / len(mn_layer2)
# 根据Mn层的z轴坐标给计算Na层的z轴坐标
Na_layer1_z = (layer1_avr_z + layer2_avr_z) / 2
Na_layer2_z = (layer1_avr_z + layer2_avr_z - 1) / 2
# 调试
print(Na_layer1_z)
print(Na_layer2_z)
# 创建最终可以插入碱金属的坐标列表
site_coord = []
# 依次按照层与z坐标对应的方法来写入坐标信息,以Mn原子的xy轴+计算得到的z轴坐标
for add_site in mn_layer1_site:
add_site = [add_site[0], add_site[1], Na_layer2_z]
site_coord.append(add_site)
for add_site in mn_layer2_site:
add_site = [add_site[0], add_site[1], Na_layer1_z]
site_coord.append(add_site)
# 调试
print(len(site_coord))
# 返回坐标信息
return site_coord
def main():
poscar = Poscar.from_file("POSCAR")
structure = poscar.structure
site_coord = get_site(structure, "Mn")
if __name__ == '__main__':
main()该代码使用了自定义函数,所以没有使用过Python的同志可能会感觉困惑,在这串代码运行时,首先会运行if __name__ == '__main__':中的内容,即运行main(),运行这行代码即运行def main():这一函数,即代码跳转到
def main():
poscar = Poscar.from_file("POSCAR")
structure = poscar.structure
site_coord = get_site(structure, "Mn")随后再运行到site_coord = get_site(structure, "Mn")时,便运行def get_site(structure, element):这一函数,即代码跳转到
def get_site(structure, element):
# 获取结构中Mn的Site信息
mn_sites = [site for site in structure if site.specie.symbol == element]
....所以这样来说,这串代码首先做的是读取文件数据,随后从文件数据中得到需要的信息,值得注意的是,该代码读取信息的部分为
poscar = Poscar.from_file("POSCAR")与我们前文的略有不同,我们是用的是Structure函数,但是这问题不大,我们使用的函数是直接读取并处理了它的结构信息,而该代码则并没有处理结构信息而是直接读取,想要得到有用的结构信息仍需要进行structure处理,即原文中的
poscar = Poscar.from_file("POSCAR")
structure = poscar.structurePoscar在某些时候是比Structure读取好的,但是在此处,我们也可以直接使用structure读取,即将main()函数改为以下内容,结果相同
def main():
structure = Structure.from_file("POSCAR")
site_coord = get_site(structure, "Mn")若将structure打印出来,可以得到以下内容
Full Formula (Mn18 O36)
Reduced Formula: MnO2
abc : 8.622101 8.622101 9.756900
angles: 90.000000 90.000000 119.999999
pbc : True True True
Sites (54)
# SP a b c
--- ---- -------- -------- ---------
0 O 0.111111 0.222222 0.099285
1 O 0.222222 0.111111 0.599285
2 O 0.222222 0.111111 0.900715
3 O 0.111111 0.222222 0.400715
4 O 0.444444 0.222222 0.099285
5 O 0.555556 0.111111 0.599285
6 O 0.555556 0.111111 0.900715
7 O 0.444444 0.222222 0.400715
8 O 0.777778 0.222222 0.099285
9 O 0.888889 0.111111 0.599285
10 O 0.888889 0.111111 0.900715
11 O 0.777778 0.222222 0.400715
12 O 0.111111 0.555556 0.099285
13 O 0.222222 0.444444 0.599285
14 O 0.222222 0.444444 0.900715
15 O 0.111111 0.555556 0.400715
16 O 0.444444 0.555556 0.099285
17 O 0.555556 0.444444 0.599285
18 O 0.555556 0.444444 0.900715
19 O 0.444444 0.555556 0.400715
20 O 0.777778 0.555556 0.099285
21 O 0.888889 0.444444 0.599285
22 O 0.888889 0.444444 0.900715
23 O 0.777778 0.555556 0.400715
24 O 0.111111 0.888889 0.099285
25 O 0.222222 0.777778 0.599285
26 O 0.222222 0.777778 0.900715
27 O 0.111111 0.888889 0.400715
28 O 0.444444 0.888889 0.099285
29 O 0.555556 0.777778 0.599285
30 O 0.555556 0.777778 0.900715
31 O 0.444444 0.888889 0.400715
32 O 0.777778 0.888889 0.099285
33 O 0.888889 0.777778 0.599285
34 O 0.888889 0.777778 0.900715
35 O 0.777778 0.888889 0.400715
36 Mn 0 0 -0
37 Mn 0 0 0.5
38 Mn 0.333333 0 -0
39 Mn 0.333333 0 0.5
40 Mn 0.666667 0 -0
41 Mn 0.666667 0 0.5
42 Mn 0 0.333333 -0
43 Mn 0 0.333333 0.5
44 Mn 0.333333 0.333333 -0
45 Mn 0.333333 0.333333 0.5
46 Mn 0.666667 0.333333 -0
47 Mn 0.666667 0.333333 0.5
48 Mn 0 0.666667 -0
49 Mn 0 0.666667 0.5
50 Mn 0.333333 0.666667 -0
51 Mn 0.333333 0.666667 0.5
52 Mn 0.666667 0.666667 -0
53 Mn 0.666667 0.666667 0.5
原代码中的各种读取信息代码便是从该内容中读取有用的信息
以下是作者的原话
接下来一步一步的再解释一下代码吧,这里我们新导入了pymatgen的Poscar类,这是vasp类下的一个分支,可以帮助我们读取POSCAR文件,所以我们在调用我们新写的get_site()函数之前,先有一个读取文件并转化为structure对象的一个过程,这个过程没有写进函数内部的原因是我们后续很多操作都需要这个structure对象,为了节约资源,避免程序冗余,所以我就将它写到了main()函数中,然后将structure对象来传递给其它函数。
随后是函数内部,其实获取Mn原子的坐标在最开始的三行代码就实现了,后面的都是为了确定我们可以直接使用的坐标列表,毕竟我们不能直接将Na插入到Mn原子的位置上,而是与Mn原子对应的层间,在常规的层状氧化物中,有一部分Na离子在层间的位置就是与Mn原子对应的,也就是你从c轴方向看xy平面,Na离子与Mn原子之间的位置是重合的,它们空间位置的差异来源于z轴坐标,所以我们这个层间的位置x和y轴坐标就可以直接参考Mn原子的x和y轴坐标,但是z轴坐标就需要重新计算得到。
这里回头来再看一下层状氧化物的模型,虽然模型上有三层,但是实际上是只有两层的Mn原子的,三层是周期性结构显示的原因:
而观察POSCAR的内容也不难发现,一部分的z轴数据在0.5附近,而另外一部分则在1.0附近,但是没有大于1的数值,反而有一些负数,这些负数其实就是周期性结构中大于1的Mn原子。所以为了方便操作我们先对这个进行了归一化处理,也就是对它们+1,对于周期性结构的分数坐标来说,这个坐标信息是等价的,然后对这两层的原子进行了分层处理,将它们分管到不同的列表中。
然后我们也就可以根据这两层的数据来计算层间Na离子的z轴坐标。计算得到合理的z轴坐标之后,就是将计算得到了两层z坐标之后,就可以将Mn原子的xy轴坐标与计算得到的z轴坐标拼接起来,凑成一个新的坐标并保存到一个列表中,这里我为了确保两层的坐标数量一致且没有坐标重复的,所以根据两层Mn来分配了z轴坐标,比如对于layer1的Mn原子的xy坐标,与它们拼接的就是计算得到的layer2_z的数据。
最后我在里面提供了三个调试的函数,就是使用print来输出数据验证我们的代码是否按照我们的想法运行了,如果说代码没有错的话,前两个print打印出来的数据应该是两个小于1的数字,而最后一个print打印的数字则应该与Mn原子的数量对应:


随后为插入Na原子
import random
def gen_structures(site_coord, structure, stuctreu_num, ele_num):
# 定义一个用来存储随机组合的坐标列表的列表
site_combine = []
for i in range(stuctreu_num):
# 随机采样三个坐标并写为列表
random_sites = random.sample(site_coord, 3)
# 保存采样的坐标
site_combine.append(random_sites)
# 存储我们需要生成的结构
new_structures = []
# 遍历之前随机产生的坐标组合列表
for random_sites in site_combine:
# 复制一个结构,方便后续进行插入操作
pre_structure = structure.copy()
# 遍历每一个坐标组合中的每个坐标
for coord in random_sites:
# 根据坐标来插入Na原子
pre_structure.insert(0, "Na", coord)
# 保存结构信息
new_structures.append(pre_structure)
#调试
print(len(new_structures))
return new_structures请注意,该内容需要嵌入上述源代码,并在main()函数中加入索引
new_structures = gen_structures(site_coord, structure, 10, 3)这部分其实已经脱离pymatgen了,只是对前文的结构内容进行修改
其中下述代码为在原本的坐标中随机获取三个 坐标并将其写入site_combine列表用于Na的插入
# 随机采样三个坐标并写为列表
random_sites = random.sample(site_coord, 3)
# 保存采样的坐标
site_combine.append(random_sites)随后将这三个坐标的元素改为Na后便可将数据传回
如果把这部分代码写的简便一点可以写为,但我觉得代码难度可能就有点高了,仁者见仁吧
def gen_structures(site_coord, structure, struct_num, ele_num):
site_combine = [random.sample(site_coord, ele_num) for _ in range(struct_num)]
new_structures = []
for sites in site_combine:
pre_structure = structure.copy()
for coord in sites:
pre_structure.insert(0, "Na", coord)
new_structures.append(pre_structure)
return new_structures随后是Eward能量的计算
from pymatgen.analysis.ewald import EwaldSummation
def get_ewald_energy(new_structures):
# 设定原子价态
element_valences = {"Na": 1, "O": -2, "Mn" : 4}
# 定义一个编号,方便编写文件
index_n = 0
# 设定文件列表
file_energy_list = []
# 遍历结构
for structure in new_structures:
index_n += 1
# 给结构中的原子添加价态
structure.add_oxidation_state_by_element(element_valences)
# 进行Ewald计算
ewald = EwaldSummation(structure)
# 求解能量
energy = ewald.total_energy
# 将编号与能量相对应
file_energy_list.append([index_n, energy])
# 按照编号给新生成的POSCAR文件命名
structure.to(fmt="poscar", filename=str(index_n) + "_POSCAR")
# 对列表按照能量大小排序,从小到大
sorted_energy_list = sorted(file_energy_list, key=lambda x: x[1])
# 遍历排序后的列表
for i in sorted_energy_list:
# 按照排好的顺序依次输出EWald能量
print("结构ID:" + str(i[0]) + " 的EWald能量为" + str(i[1]))和Na原子插入一样,也是需要嵌入源代码然后在main函数写入
get_ewald_energy(new_structures)以下为作者原话
要求解Ewald能量就需要用到pymatgen中的EwaldSummation类,所以需要导入这个类,然后将之前得到的new_structures列表传递给函数,函数内遍历这个列表,获取每个结构,给每个结构添加价态,然后就是求解Ewald能量,求解之后按照编号+能量的方法来组成一个新的列表并将每个结构输出为POSCAR文件,最后还会打印每个结构的Ewald能量。
到了这一步,运行之后的结果如下:
中间有一些warning,这是无害的,如果看不惯可以考虑导入warning库来忽略这些警报,同时这个脚本所在的目录下也会多出来10个POSCAR格式的文件,并且按照1-10的编号命名好了的,可以自行查看。
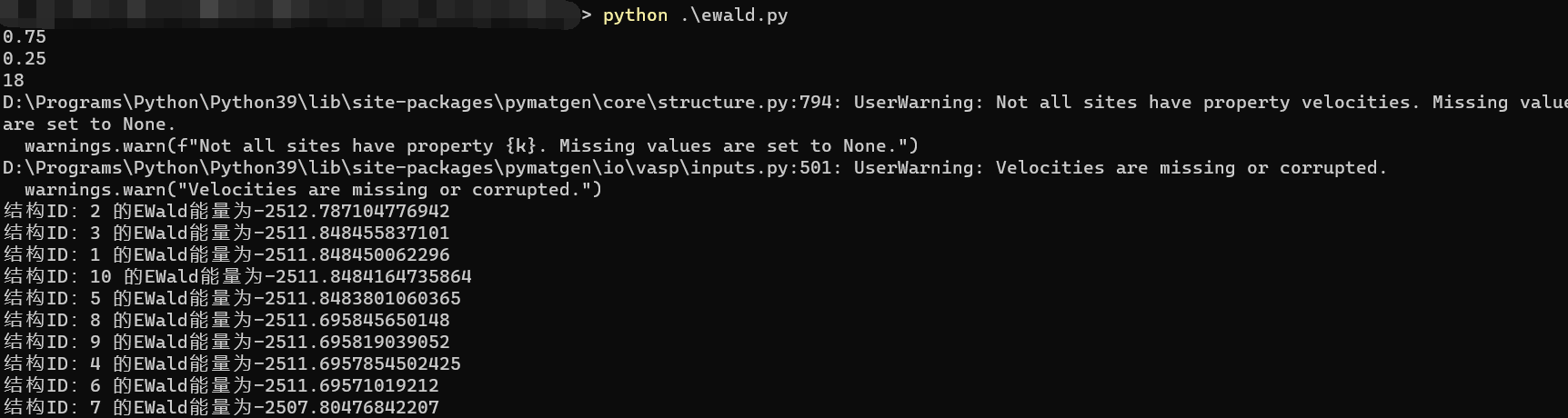
整理后便可以得到完整的代码
from pymatgen.io.vasp import Poscar
import random
from pymatgen.analysis.ewald import EwaldSummation
def get_site(structure, element):
# 获取结构中Mn的Site信息
mn_sites = [site for site in structure if site.specie.symbol == "Mn"]
# 获取结构中Mn的坐标信息
mn_coords = [site.frac_coords for site in mn_sites]
# 获取结构中Mn的z轴坐标
mn_z_coords = [site.frac_coords[2] for site in mn_sites]
# 对结构中Mn的z轴坐标进行归一处理
mn_z_coords = [1 + i if i <= 0.25 else i for i in mn_z_coords]
# 对结构中Mn原子进行分层处理
mn_layer1 = [i for i in mn_z_coords if i > 0.75 ]
mn_layer2 = [i for i in mn_z_coords if i <= 0.75 ]
# 按照层将每层Mn原子的z轴坐标提取出来
mn_layer1_site = [mn_coords[i] for i, z_coord in enumerate(mn_z_coords) if z_coord > 0.75]
mn_layer2_site = [mn_coords[i] for i, z_coord in enumerate(mn_z_coords) if z_coord <= 0.75]
# 求解Mn层的平均z轴坐标
layer1_avr_z = sum(mn_layer1) / len(mn_layer1)
layer2_avr_z = sum(mn_layer2) / len(mn_layer2)
# 根据Mn层的z轴坐标给计算Na层的z轴坐标
Na_layer1_z = (layer1_avr_z + layer2_avr_z) / 2
Na_layer2_z = (layer1_avr_z + layer2_avr_z - 1) / 2
# 调试
print(Na_layer1_z)
print(Na_layer2_z)
# 创建最终可以插入碱金属的坐标列表
site_coord = []
# 依次按照层与z坐标对应的方法来写入坐标信息,以Mn原子的xy轴+计算得到的z轴坐标
for add_site in mn_layer1_site:
add_site = [add_site[0], add_site[1], Na_layer2_z]
site_coord.append(add_site)
for add_site in mn_layer2_site:
add_site = [add_site[0], add_site[1], Na_layer1_z]
site_coord.append(add_site)
# 调试
print(len(site_coord))
# 返回坐标信息
return site_coord
def gen_structures(site_coord, structure, stuctreu_num, ele_num):
# 定义一个用来存储随机组合的坐标列表的列表
site_combine = []
for i in range(stuctreu_num):
# 随机采样三个坐标并写为列表
random_sites = random.sample(site_coord, 3)
# 保存采样的坐标
site_combine.append(random_sites)
# 存储我们需要生成的结构
new_structures = []
# 遍历之前随机产生的坐标组合列表
for random_sites in site_combine:
# 复制一个结构,方便后续进行插入操作
pre_structure = structure.copy()
# 遍历每一个坐标组合中的每个坐标
for coord in random_sites:
# 根据坐标来插入Na原子
pre_structure.insert(0, "Na", coord)
# 保存结构信息
new_structures.append(pre_structure)
#调试
# print(len(new_structures))
return new_structures
def get_ewald_energy(new_structures):
# 设定原子价态
element_valences = {"Na": 1, "O": -2, "Mn" : 4}
# 定义一个编号,方便编写文件
index_n = 0
# 设定文件列表
file_energy_list = []
# 遍历结构
for structure in new_structures:
index_n += 1
# 给结构中的原子添加价态
structure.add_oxidation_state_by_element(element_valences)
# 进行Ewald计算
ewald = EwaldSummation(structure)
# 求解能量
energy = ewald.total_energy
# 将编号与能量相对应
file_energy_list.append([index_n, energy])
# 按照编号给新生成的POSCAR文件命名
structure.to(fmt="poscar", filename=str(index_n) + "_POSCAR")
# 对列表按照能量大小排序,从小到大
sorted_energy_list = sorted(file_energy_list, key=lambda x: x[1])
# 遍历排序后的列表
for i in sorted_energy_list:
# 按照排好的顺序依次输出EWald能量
print("结构ID:" + str(i[0]) + " 的EWald能量为" + str(i[1]))
def main():
# 读取文件并转化为structure
filename = "POSCAR"
poscar = Poscar.from_file(filename)
structure = poscar.structure
site_coord = get_site(structure, "Mn")
new_structures = gen_structures(site_coord, structure, 10, 3)
get_ewald_energy(new_structures)
if __name__ == '__main__':
main()
开放原子开发者工作坊旨在鼓励更多人参与开源活动,与志同道合的开发者们相互交流开发经验、分享开发心得、获取前沿技术趋势。工作坊有多种形式的开发者活动,如meetup、训练营等,主打技术交流,干货满满,真诚地邀请各位开发者共同参与!
更多推荐
 27
27 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)