数据的质量控制软件----fastQC
FastQC的基本介绍:FastQC是一款基于Java的软件,它可以快速地对测序数据进行质量评估,其官网为:Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data高通量测序数据的高级质控工具输入FastQ,SAM,BAM文件,输出对测序数据评估的网页报告
一、前言
FastQC的基本介绍:
FastQC是一款基于Java的软件,它可以快速地对测序数据进行质量评估,其官网为:Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data
- 高通量测序数据的高级质控工具
- 输入FastQ,SAM,BAM文件,输出对测序数据评估的网页报告
二、安装
这里需要安装Conda (这是一款用于安装多数生物信息分析软件的管理软件,重要的是可以解决软件依赖问题)
conda install fastqc三、使用fastqc
1.介绍fastqc
我们在服务器上用命令行来运行fastQC:
fastqc -t 12 -o out_path sample1_1.fq sample1_2.fq运行结束后生成两个文件一个.html网页文件,一个是.zip压缩文件。

常用参数:
-o 用来指定输出文件的所在目录,生成的报告的文件名是根据输入来定的,注意是不能自动新建目录的。输出的结果是.zip文件,默认不解压缩,命令里加上--extract则压缩。
-f 用来强制指定输入文件格式,默认自动检测。支持fastq、bam、sam极相应的gz压缩格式
-c 污染物选项,输入的是一个文件,格式是Name[Tab] Sequence,#开头的行是注释,里面是可能的污染序列,如果有这个选项,FastQC会在计算时候评估污染的情况,并在统计的时候进行分析。
-q 会进入沉默模式,指定这个选项的时候,程序不会实时报告运行的状况,即不出现下面的提示:

1.参数大小写敏感
2.参数两种形式
• 长参数
• 短参数
【更详细的使用说明书可以看官网哦】
Index of /projects/fastqc/Help http://www.bioinformatics.babraham.ac.uk/projects/fastqc/Help/
http://www.bioinformatics.babraham.ac.uk/projects/fastqc/Help/
2.运行数据质控fastqc
目标:使用fastqc对原始数据进行质量评估
```bash
# 激活conda环境
conda activate rna
# 连接数据到自己的文件夹
cd $HOME/project/Human-16-Asthma-Trans/data/rawdata
ln -s /home/t_rna/data/airway/fastq_raw25000/*gz ./
# 使用FastQC软件对单个fastq文件进行,结果输出到qc/文件夹下
nohup fastqc -t 6 -o ./ SRR*.fastq.gz >qc.log &
# 使用MultiQc整合FastQC结果
# 使用绝对路径运行
multiqc=/home/t_rna/miniconda3/envs/rna/bin/multiqc
fastqc=/home/t_rna/miniconda3/envs/rna/bin/fastqc
fq_dir=$HOME/project/Human-16-Asthma-Trans/data/rawdata
outdir=$HOME/project/Human-16-Asthma-Trans/data/rawdata
# 使用绝对路径运行
$fastqc -t 6 -o $outdir ${fq_dir}/SRR*.fastq.gz >${fq_dir}/qc.log
# 报告整合
$multiqc $outdir/*.zip -o $outdir/ >${fq_dir}/multiqc.log
```【上面的代码可以具体看看,然后在服务器上运行一下】


【我的文件比较大,大家在实战的时候也会是这样的,运行的时间可能会稍微长一点】

【运行成功的结果是这样的↑】
(多了两种文件,一种是.html,另一种是.zip文件)

【然后就可以在本地浏览器上打开啦】
四、数据质控-fastqc运行
脚本后台运行:nohup &
• nohup:no hang up(不挂起),退出终端不会影响程序的运行
• &:后台运行

【注意:./ 后面是有个空格哦】
五、 fastQC报告解读
1.Basic Statistics基本统计
【我使用的是真实数据,所以文件比较大一点】


【%GC 是我们需要重点关注的一个指标,这个值表示的是全部序列中的GC含量,这个数值一般是物种特异的,比如人类基因组就是42%左右。】
Encoding指测序平台的版本和相应的编码版本号,可推测是Phred 33 或是Phred 64 质量分数的编码方式。
Total Sequences输入文本的reads的数量。
Sequence length 测序的长度
注意:↓

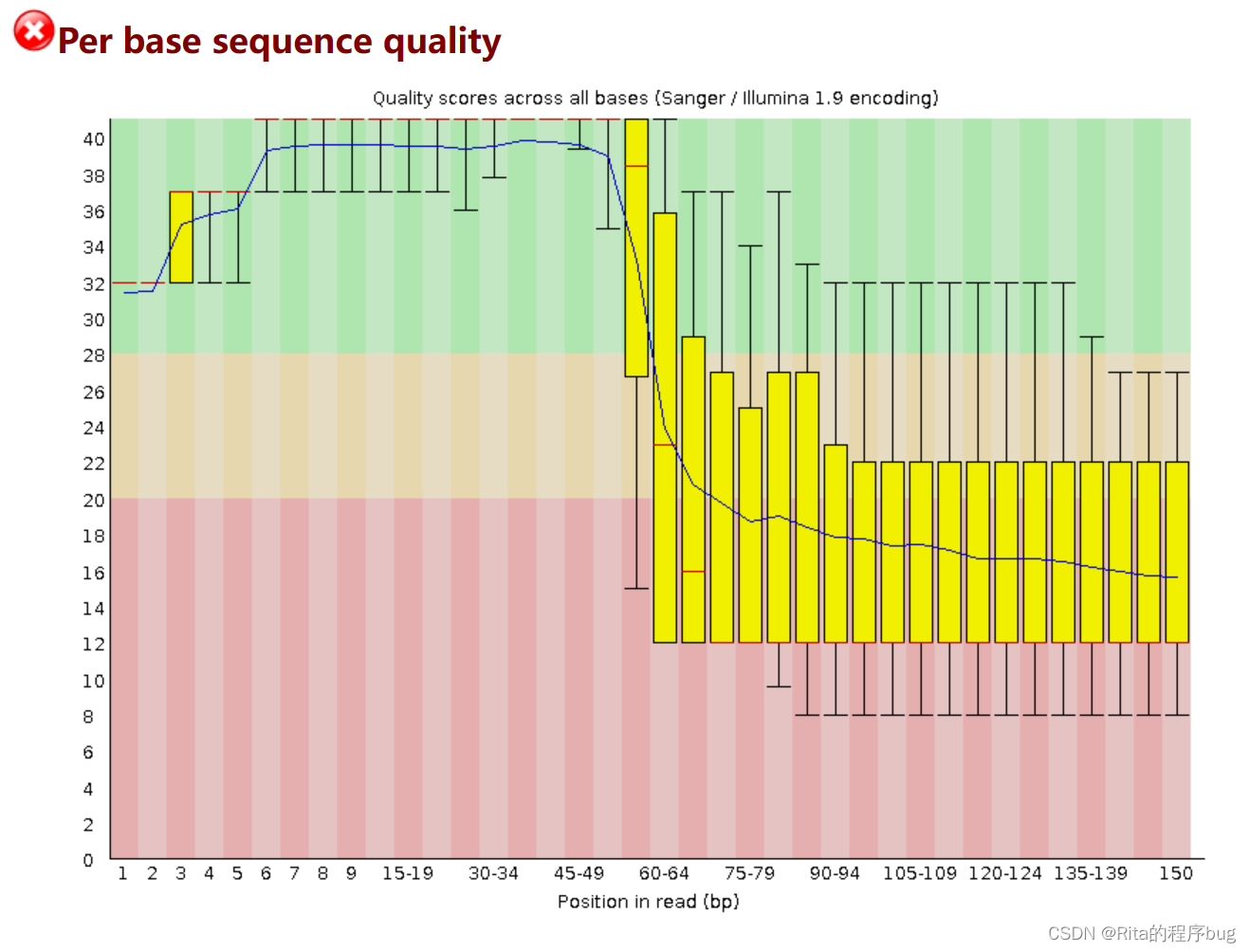
2.Per base sequence quality每个碱基序列的质量

这幅图展示的是fq文件中每一个位置上的所有read的碱基质量值的一个箱式图。 比如,我的fq有96612385个read,第一个位置上的base(碱基)就有96612385个质量值,这个值画成第一个箱 子,依此往后推。
用箱式图的方式展示数据质量,图中X轴每1个位置,都是该位置的所有序列的测序质量的统计。纵轴是质量得分,Q =-10*log10(p),p为测错的概率。所以一条reads某位置出错概率0.01时,其quality就是20。横轴是测序序列的位置。一般要求此图中,所有位置的10%分位数大于20,也就是常说的Q20过滤。
上面的须须和下面的须须分别表示10%和90%
盒子的界限为上分位数和下分位数
图中红线表示中值
图中蓝色的细线是各个位置的平均值的连线
- 根据quality给出质量结果:正常区间(28 - 40),警告区间(20-28),错误区间(0-20)。
- 比如,当read的某一位置的p=0.01,quality=20,那么它就处于错误区间。
所以上面的这个测序结果质量不好。如果任何碱基质量低于10,或者是任何中位数低于25报警,如果任何碱基质量低于5,或者是任何中位数低于20报错。
2.1为什么随着测序读长变长,碱基质量值下降?

随着测序读长变长,碱基质量值下降的原因主要有两个方面:
-
测序错误累积:在测序过程中,每个碱基都需要被准确地识别和记录。然而,随着读长的增加,测序错误的累积也会增加。这是因为在较长的读长中,测序机器可能会出现更多的错误,例如碱基识别错误、插入或删除错误等。这些错误会导致碱基质量值下降。
-
碱基信号衰减:在测序过程中,测序仪器会通过检测碱基发出的荧光信号来确定碱基的序列。然而,随着读长的增加,碱基发出的信号会逐渐衰减。这是因为荧光信号在传输过程中会受到衰减和干扰,导致信号强度减弱。因此,较长的读长会导致碱基信号变弱,进而影响到碱基质量值的准确性。
3.Per tile sequence quality每个reads序列的质量

这一模块是检查在测序平台上,reads中每一个碱基位置在不同的测序小孔之间的偏离度,偏离度越高,碱基质量越差。纵轴表示测序小孔,蓝色表示低于平均偏离度,越红则说明偏离平均质量方差越多,也就是说质量越差,本图中都是蓝色表明质量很好。如果出现质量问题可能是短暂的,如有气泡产生,也可能是长期的,如在某一小孔中存在杂质。偏离度小于平均值2以上报警,偏离度小于平均值5以上不合格。

【Bad quality↑】
4.Per sequence quality scores每个序列的质量分数

这是为了检测一部分质量特别差的reads,如果有则会在图上出现多个峰,如在测序仪边缘的reads。纵轴是reads数目,横轴是质量分数,代表不同Phred值对应了多少的reads。
本图中,测序结果主要集中在高分中,证明测序质量良好。当峰值小于27(错误率0.2%)时警报,当峰值小于20(错误率1%)时不合格。
【我的这个样本测序质量不是很好,因为它的峰值小于27,所以它会出现警报】

5.Per base sequence content

统计reads每个位置ATCG四种碱基的分布:
横轴为碱基位置,纵轴为百分比。
因为随机的文库中,正常情况下所有位置出现某种碱基的概率是相近的,因此好的测序结果中四条线应该平行且接近。
当部分位置碱基的比例出现bias时,即四条线在某些位置纷乱交织,往往提示我们有overrepresented sequence的污染。
当所有位置的碱基比例一致的表现出bias时,即四条线平行但分开,往往代表文库有bias (建库过程或本身特点),或者是测序中的系统误差。
当任一位置的A/T比例与G/C比例相差超过10%,报"WARN";
当任一位置的A/T比例与G/C比例相差超过20%,报"FAIL"。
6.Per sequence GC content每个序列GC含量

每个碱基位置上:ATGC含量的分布图
横轴:各碱基位置;
纵轴:碱基百分比
统计reads的平均GC含量分布
红线是实际情况,蓝线是理论分布(正态分布,均值不一定在50%,而是由平均GC含量推断的)。 曲线形状的偏差往往是由于文库的污染或是部分reads构成的子集有偏差(overrepresented reads)。形状接近正态但偏离理论分布的情况提示我们可能有系统偏差。
偏离理论分布的reads超过15%时,报"WARN";偏离理论分布的reads超过30%时,报"FAIL"。
7.Per base N content每基N含量

统计reads每个位置N的比率
reads某个位置无法确定是何种碱基时,使用N代替;
正常情况下,N的比例是很小的,所以图上常常看到一条直线,但放大Y轴之后会发现还是有N的存在,这不算问题。当Y轴在0%-100%的范围内也能看到“鼓包”时,说明测序系统出了问题。
当任意位置的N的比例超过5%,报"WARN";
当任意位置的N的比例超过20%,报"FAIL"。
8.Sequence Length Distribution序列长度分布

reads长度分布
为了防止建库或者测序时有一些不规则长度的序列也被进行测序而进行的一个对长度的统计,当所有序列的长度不一样,fastqc就会警告。
当reads长度不一致时报"WARN";
当有长度为0的read时报“FAIL”。
【比如此图中,150bp是主要的,但是还是有少量的149和151bp的长度】
每次测序仪测出来的长度在理论上应该是完全相等的。
当测序的长度不同时,如果很严重,则表明测序仪在此次测序不成功
9.Sequence Duplication Levels序列重复级别

统计reads重复水平
测序本身就会产生重复reads,测序深度越高,reads重复数越大;如果重复出现峰值,就提示可能b存在偏差(如建库过程中的PCR duplication)。
横坐标是重复的次数,纵坐标是duplicated reads占unique reads种数百分比。
fastqc抽取reads文件前200,000条reads统计其重复情况。重复数目大于等于10的reads被合并统计,这也是为什么我们看到上图的最右侧略有上扬。大于75bp的reads只取50bp进行比较。由于reads越长错误率越高,所以其重复程度仍有可能被低估。
当非unique的reads占总数的比例大于20%时,报"WARN";
当非unique的reads占总数的比例大于50%时,报"FAIL“。
10.Overrepresented sequences代表性过高的序列
过度重复出现的序列的统计信息(此次没有)
11.Adapter Content

Adapter序列在reads中出现概率
接头含量:接头序列统计,>5%时是Warning,>10%时是Failure。
表示序列中两端adapter的情况 软件内置了四种常用的测序接头序列, fastqc 有一个参 数-a可以自定义接头序列
12.质量好的样本vs质量不好的样本
作为初学者,也许你并不知道好的样本fastqc报告结果是怎样的,所以官网提供了对比例子。


开放原子开发者工作坊旨在鼓励更多人参与开源活动,与志同道合的开发者们相互交流开发经验、分享开发心得、获取前沿技术趋势。工作坊有多种形式的开发者活动,如meetup、训练营等,主打技术交流,干货满满,真诚地邀请各位开发者共同参与!
更多推荐
 68
68 0
0- 0



 已为社区贡献2条内容
已为社区贡献2条内容

所有评论(0)