RNA 8. SCI文章中差异基因表达--热图 (heatmap)
大多我们在做完差异表达之后都会看下我们的差异基因筛选的是否能将分组结果展现出来,都会选择热图,主要是热图技能聚类,又可以展现表达量的大小,非常直观,所以这期我们就说下热图的绘制方法。
前言
大多我们在做完差异表达之后都会看下我们的差异基因筛选的是否能将分组结果展现出来,都会选择热图,主要是热图技能聚类,又可以展现表达量的大小,非常直观,所以这期我们就说下热图的绘制方法。
实例解析
1. 数据读取
数据的读取我们仍然使用的是 TCGA-COAD 的数据集,表达数据的读取以及临床信息分组的获得我们上期已经提过,我们使用的是edgeR 软件包计算出来的差异表达结果,合并了原始的 Count 数据,如下:
DEG=read.table("DEG-resdata.xls",sep="\t",check.names=F,header = T)
PCAmat<-DEG[,8:ncol(DEG)]
rownames(PCAmat)=DEG[,1]
PCAmat[1:5,1:3]
## TCGA-3L-AA1B-01A-11R-A37K-07 TCGA-4N-A93T-01A-11R-A37K-07
## ENSG00000142959 20 15
## ENSG00000163815 175 108
## ENSG00000107611 49 13
## ENSG00000162461 55 89
## ENSG00000163959 153 259
## TCGA-4T-AA8H-01A-11R-A41B-07
## ENSG00000142959 49
## ENSG00000163815 59
## ENSG00000107611 6
## ENSG00000162461 48
## ENSG00000163959 102
读取分组信息癌组织样本 478 和癌旁组织样本 41,如下:
group<-read.table("DEG-group.xls",sep="\t",check.names=F,header = T)
head(group)
## Sample Group
## 1 TCGA-D5-6530-01A-11R-1723-07 TP
## 2 TCGA-G4-6320-01A-11R-1723-07 TP
## 3 TCGA-AD-6888-01A-11R-1928-07 TP
## 4 TCGA-CK-6747-01A-11R-1839-07 TP
## 5 TCGA-AA-3975-01A-01R-1022-07 TP
## 6 TCGA-A6-6780-01A-11R-1839-07 TP
table(group$Group)
##
## NT TP
## 41 478
2. Count 转换为 FPKM
我们之前下载的数据是 Count 用来做差异表达分析,而在绘制热图时需要我们使用RPKM或者时FPKM,那么我们利用差异基因的结果,提取对应的基因,得到Count 矩阵,下载基因长度,来计算FPKM即可,还记得我们之前讲过的FPKM的计算公式,
FPKM (Fragments Per Kilobase of exon model per Million mapped fragments)
-
FPKM:每千个碱基的转录每百万映射读取的fragments,主要是针对pair-end测序表达量进行计算。
-
FPKM和RPKM的区别就是一个是fragment,一个是read。
那么我们首先下载外显子的注释信息,GDC Reference Files | NCI Genomic Data Commons (cancer.gov) 找到如下信息,下载即可。
外显子注释文件的处理,我们希望得到第一列时基因,第二列时基因长度。
安装并加载软件包 GenomicFeatures,这个软件包在处理注释文件时非常方便,如下:
if(!require(GenomicFeatures)){
BiocManager::install("GenomicFeatures")
}
library(GenomicFeatures)
读取下载的注释文件,并获取外显子的总长度,如下:
txdb <- makeTxDbFromGFF("gencode.v22.annotation.gtf.gz",format = "gtf")
exons <- exonsBy(txdb,by="gene")
exons_length<-lapply(exons,function(x){sum(width(reduce(x)))})
class(exons_length)
## [1] "list"
length(exons_length)
## [1] 60483
exons_length<-as.data.frame(exons_length)
dim(exons_length)
## [1] 1 60483
exons_length1<-t(exons_length)
exons_length1<-as.data.frame(exons_length1)
dim(exons_length1)
## [1] 60483 1
head(exons_length1)
## V1
## ENSG00000000003.13 4535
## ENSG00000000005.5 1610
## ENSG00000000419.11 1207
## ENSG00000000457.12 6883
## ENSG00000000460.15 5967
## ENSG00000000938.11 3474
我们发现差异基因后面时不带 “.” 和数字的,所以需要我们去掉基因后面的数字,如下:
colnames(exons_length1)="Length"
Gene<-gsub("\\.(\\.?\\d*)","",rownames(exons_length1))
exons_length1$Gene=as.data.frame(Gene)[,1]
head(exons_length1)
## Length Gene
## ENSG00000000003.13 4535 ENSG00000000003
## ENSG00000000005.5 1610 ENSG00000000005
## ENSG00000000419.11 1207 ENSG00000000419
## ENSG00000000457.12 6883 ENSG00000000457
## ENSG00000000460.15 5967 ENSG00000000460
## ENSG00000000938.11 3474 ENSG00000000938
根据提取差异基因并与 Count 矩阵合并,矩阵最后一列时基因长度,如下:
PCAmat$Gene<-rownames(PCAmat)
####count 矩阵,长度合并
count_length<-merge(PCAmat,exons_length1,by="Gene")
计算 FPKM,利用合并后的结果,样本有519个,那么就是从2列到520列,结合公式开始计算 FPKM,如下:
kb<-count_length$Length/1000
countdata<-count_length[,2:520]
rpk <- countdata/kb
fpkm <-t(t(rpk)/colSums(countdata) * 10^6)
rownames(fpkm)=rownames(PCAmat)
fpkm[1:5,1:3]
## TCGA-3L-AA1B-01A-11R-A37K-07 TCGA-4N-A93T-01A-11R-A37K-07
## ENSG00000142959 1.5336078 1.826404
## ENSG00000163815 51.1904250 388.921103
## ENSG00000107611 162.7697536 52.262338
## ENSG00000162461 69.9931252 93.087394
## ENSG00000163959 0.2628356 0.000000
## TCGA-4T-AA8H-01A-11R-A41B-07
## ENSG00000142959 0.7546987
## ENSG00000163815 178.9371541
## ENSG00000107611 13.3500151
## ENSG00000162461 90.5084641
## ENSG00000163959 0.0000000
FPKM 数据进行标准,如下:
data=log2(fpkm+1)
dat=t(scale(t(data))) # 'scale'可以对log(fpkm+1)数值进行归一化
#处理数据
dat[dat>2]=2
dat[dat<(-2)]= -2
dim(dat)
## [1] 4128 519
dat[1:5,1:3]
## TCGA-3L-AA1B-01A-11R-A37K-07 TCGA-4N-A93T-01A-11R-A37K-07
## ENSG00000142959 0.312929749 0.51326990
## ENSG00000163815 -0.451067532 1.30683599
## ENSG00000107611 0.811371990 0.01667015
## ENSG00000162461 -0.076316888 0.23114035
## ENSG00000163959 -0.004643292 -0.62802639
## TCGA-4T-AA8H-01A-11R-A41B-07
## ENSG00000142959 -0.3600191
## ENSG00000163815 0.6308436
## ENSG00000107611 -0.9112186
## ENSG00000162461 0.2008002
## ENSG00000163959 -0.6280264
3. 绘制热图
热图绘制使用软件包 pheatmap,这个软件包使用起来非常方便,参数多能够满足各种需求,更重要的是提取聚类数据也可以自己二次处理聚类结果,注释方便。
安装并加载软件包,如下:
if(!require(pheatmap)){
install.packages("pheatmap")
}
library(pheatmap)
最简单的一种方式绘制热图,如下:
###########pheatmap
#调整参数,换颜色
pheatmap(dat,
cluster_rows = TRUE,
show_rownames=FALSE,
show_colnames = FALSE,
scale="none",
cluster_cols = F,
fontsize_row = 10,
fontsize_col = 10,
#color = colorRampPalette(c("navy", "white", "firebrick3"))(100),
color = colorRampPalette(c("green3", "white", "blue4"))(100),#换颜色
angle_col = 45, #修改横轴坐标名倾斜度
filename = 'cor.fpkm1.png',
)

添加加注释信息,需要对分组信息做一下出来,
首先是样本的分组注释,根据癌症组和正常组整理,也就是列注释数据处理,如下:
#######加注释信息
rownames(group)=group$Sample
head(group)
## Sample Group
## TCGA-D5-6530-01A-11R-1723-07 TCGA-D5-6530-01A-11R-1723-07 TP
## TCGA-G4-6320-01A-11R-1723-07 TCGA-G4-6320-01A-11R-1723-07 TP
## TCGA-AD-6888-01A-11R-1928-07 TCGA-AD-6888-01A-11R-1928-07 TP
## TCGA-CK-6747-01A-11R-1839-07 TCGA-CK-6747-01A-11R-1839-07 TP
## TCGA-AA-3975-01A-01R-1022-07 TCGA-AA-3975-01A-01R-1022-07 TP
## TCGA-A6-6780-01A-11R-1839-07 TCGA-A6-6780-01A-11R-1839-07 TP
group1=group[colnames(dat),-1,drop=FALSE]
head(group1)
## Group
## TCGA-3L-AA1B-01A-11R-A37K-07 TP
## TCGA-4N-A93T-01A-11R-A37K-07 TP
## TCGA-4T-AA8H-01A-11R-A41B-07 TP
## TCGA-5M-AAT4-01A-11R-A41B-07 TP
## TCGA-5M-AAT5-01A-21R-A41B-07 TP
## TCGA-5M-AAT6-01A-11R-A41B-07 TP
加入列注释,再进行绘制热图,如下:
pheatmap(dat,
cluster_rows = FALSE,
show_rownames=FALSE,
show_colnames = FALSE,
scale="none",
cluster_cols = FALSE,
fontsize_row = 10,
fontsize_col = 10,
annotation_col = group1,
color = colorRampPalette(c("navy", "white", "firebrick3"))(100),
#color = colorRampPalette(c("green3", "white", "blue4"))(100),#换颜色
angle_col = 45, #修改横轴坐标名倾斜度
filename = 'cor.fpkm2.png',
)

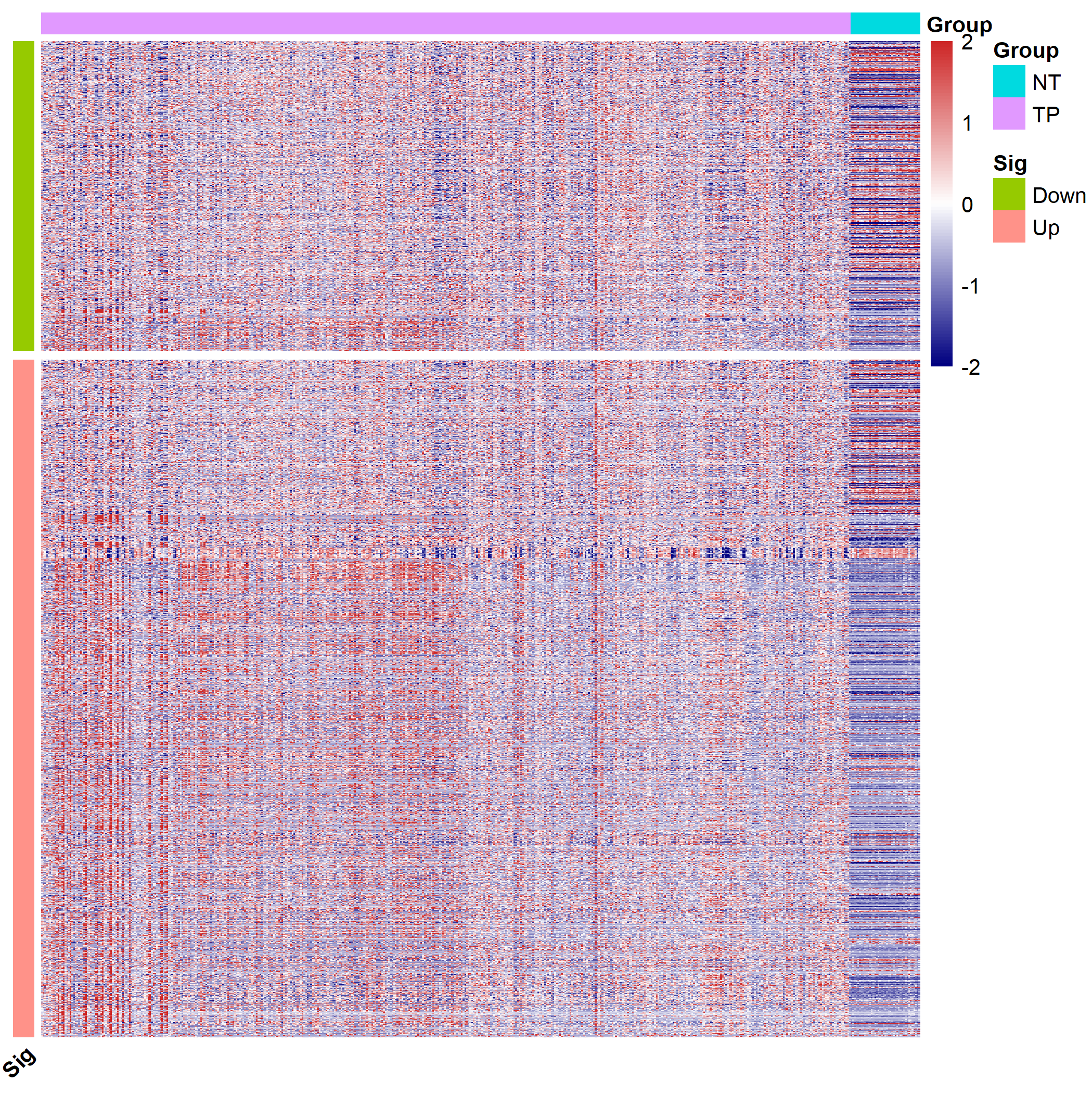
根据差异结果中 Up, Down 分组来调整基因的顺序,在对行进行处理,这其实就是对基因的一个分组,分为Up 和 Down,如下:
ann_row=as.data.frame(DEG[,c(1,7)])
colnames(ann_row)=c("Sample","Sig")
rownames(ann_row)=DEG$Row.names
ann_row=ann_row[order(ann_row$Sig),]
rownames(ann_row)=ann_row$Sample
ann_row=as.data.frame(ann_row)
ann_row1=as.data.frame(ann_row[,2])
rownames(ann_row1)=ann_row$Sample
colnames(ann_row1)="Sig"
head(ann_row1)
## Sig
## ENSG00000142959 Down
## ENSG00000163815 Down
## ENSG00000107611 Down
## ENSG00000162461 Down
## ENSG00000163959 Down
## ENSG00000144410 Down
加入行列注释,再进行绘制热图,如下:
#######根据差异结果中 Up, Down 分组来调整基因的顺序
pheatmap(dat[rownames(ann_row1),],
cluster_rows = FALSE,
show_rownames=FALSE,
show_colnames = FALSE,
scale="none",
cluster_cols = FALSE,
fontsize_row = 10,
fontsize_col = 10,
annotation_col = group1,
annotation_row = ann_row1,
color = colorRampPalette(c("navy", "white", "firebrick3"))(100),
#color = colorRampPalette(c("green3", "white", "blue4"))(100),#换颜色
angle_col = 45, #修改横轴坐标名倾斜度
filename = 'cor.fpkm3.png',
)

拆分多个模块,我们先看下基因上下调个数,下调基因数为1296,如下
table(ann_row1$Sig)
##
## Down Up
## 1296 2832
根据下调基因的个数,设置 gaps_row=1296,如下:
#########拆分多个模块
pheatmap(dat[rownames(ann_row1),],
cluster_rows = FALSE,
show_rownames=FALSE,
show_colnames = FALSE,
scale="none",
cluster_cols = FALSE,
fontsize_row = 10,
fontsize_col = 10,
annotation_col = group1,
annotation_row = ann_row1,
color = colorRampPalette(c("navy", "white", "firebrick3"))(100),
#color = colorRampPalette(c("green3", "white", "blue4"))(100),#换颜色
angle_col = 45, #修改横轴坐标名倾斜度
filename = 'cor.fpkm4.png',
gaps_row=1296#拆分位置
)

完成,非常简单,但是也需要大家仔细认真地把数据准备好,关注公众号,扫码进群,精彩内容不断更新!

桓峰基因
生物信息分析,SCI文章撰写及生物信息基础知识学习:R语言学习,perl基础编程,linux系统命令,Python遇见更好的你
45篇原创内容
公众号

开放原子开发者工作坊旨在鼓励更多人参与开源活动,与志同道合的开发者们相互交流开发经验、分享开发心得、获取前沿技术趋势。工作坊有多种形式的开发者活动,如meetup、训练营等,主打技术交流,干货满满,真诚地邀请各位开发者共同参与!
更多推荐
 1
1 0
0- 0



 已为社区贡献2条内容
已为社区贡献2条内容

所有评论(0)