orthofinder直系同源蛋白分析及结果处理
文章目录安装使用简介摘要背景介绍安装(gtdbtk) [yutao@myosin Genome_integration]$ mamba create -n orthofinder -c bioconda orthofinder使用简介OrthoFinder: phylogenetic orthology inference for comparative genomicsGenome Biolog
文章目录
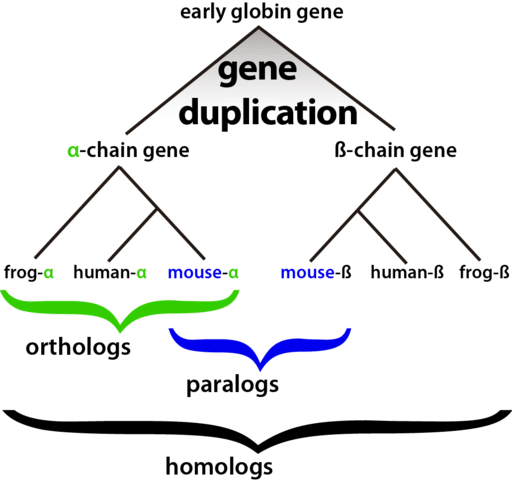
同源概念


引自:知乎:直系同源(Orthologs)和旁系同源(Paralogs)
安装
(gtdbtk) [yutao@myosin Genome_integration]$ mamba create -n orthofinder -c bioconda orthofinder
使用
(orthofinder) [yutao@myosin 137MIMAG_High_quality_MAGs]$ ulimit -n 20000 # 临时设置最大文件打开量
nohup time orthofinder -f test -t 100 -a 100 -o test_orthofinder &>of.log&
# -t 是blast all2all的线程
# -a 是做后续步骤的线程
- test是包含蛋白序列的目录,每个基因组一个文件
详细输出结果
对结果的初步了解
默认情况下,OrthoFinder会在输入的蛋白质组目录下创建一个名为 "OrthoFinder "的结果目录,并将结果放在这里。我的结果目录看起来像这样。

质量控制:Percentage of genes in orthogroups
我喜欢检查的第一件事是有多少基因被分配到orthogroups。在OrthoFinder的输出中,Comparative_Genomics_Statistics/Statistics_Overall.tsv,它打印了这样的文字。
OrthoFinder assigned 121743 genes (92.9% of total) to 17981 orthogroups.
这是非常好的,一般来说,看到你至少有80%的基因被分配到orthogroups是很好的。比这少意味着你可能遗漏了一些剩余基因实际存在的orthogroups关系,不良的物种抽样是最可能的原因。我们也来检查一下每个物种的百分比。有一个名为Comparative_Genomics_Statistics/Statistics_PerSpecies.tsv的标签分隔文件。与OrthoFinder的其他".tsv "文件一样,这个文件最好在Excel或LibreOffice Calc等电子表格程序中查看。这些文件在你的电脑上可能会被自动正确处理,或者你可能需要明确告诉它它们是以制表符分隔的。以下是你如何用LibreOffice或Excel进行处理。

一旦你打开这个文件,你会看到脊椎动物都有超过90%的基因被分配到OG,而果蝇有大约76%的基因被分配到OG。这可能是由于物种抽样的原因。四个脊椎动物物种都是相对密切相关的,而果蝇和秀丽隐杆线虫的物种抽样都很差。为了改善这种情况,我们需要包括一些物种,以打破物种树中把这些物种与所有其他物种分开的长枝。
物种树
接下来让我们看看物种树。Dendroscope是一个可以下载并在本地运行的树查看器,如果你要查看超过几棵树,它是最好的选择。另外,也有一些选项可以从你的网络浏览器中运行,例如ETE工具包的树查看器。使用其中之一,打开Species_Tree/SpeciesTree_rooted.txt文件。由于这个文件有引导值,Dendroscope需要你选择 "解释为边缘标签 "的选项来正确查看它们。该物种树看起来像这样。

这棵树是由OrthoFinder使用STAG算法推断出来的,并使用STRIDE算法进行了根化,因此可以随时进行解读(通常情况下,你必须先对一棵树进行根化)。你可以在这里看到果蝇的分支比其他物种长,如上所述。如果你知道物种树应该是什么样子的,你应该检查一下树是否与你所期望的一致。OrthoFinder在这里推断出的树是正确的。
如果物种树不正确,那么这不会影响OG的推断,但是可能会影响到一些有基因复制事件的基因树的OG推断。在这种情况下,你可能想用校正后的物种树再次运行OrthoFinder分析的最后一点(-ft和-s选项)。这通常是非常快的,因为所有的计算费用(正交群和基因树的推断等)都已经完成了。参见OrthoFinder的最佳实践。

137MAGs Orthofinder推断的物种树
顺便提一下。你会注意到在这棵树上,支持率值并不都是100%,而你可能期望它们在这个数据集中是100%。在默认选项下,物种树推断是用STAG进行的,它使用从单焦点基因树中得到的支持每个双区的物种树的比例作为其支持度的测量。这是一个比来自多序列排列的标准自举支持度更严格的衡量标准。如果使用了"-M msa "选项,那么在推断物种树时就会使用串联的多序列排列,所有双区的支持率就会达到100%。在这种情况下,支持值对应于从完整的多基因排列中提取的自举复制,这完全是两回事。这是最常见的支持度测量方法,对于相同的数据,总是会报告更高的支持度值。
Orthologues
运行OrthoFinder最常见的原因之一是寻找你感兴趣的基因的orthologue。让我们来看看果蝇基因FBgn0005648的orthologue,它参与核裂解/聚腺苷酸化反应的裂解和聚腺苷酸化步骤(见FlyBase)。这是一个有趣的基因,因为在通向人类的血统中发生了两次基因复制事件,我们将看到这一点。让我们来看看它在人类中的同源物是什么。
在Orthologues目录中,每个物种都有一个子目录。在电子表格程序中打开Orthologues/Orthologues_Drosophila_melanogaster/Drosophila_melanogaster__v__Homo_sapiens.tsv(必要时指定为制表符分隔)。该文件有三栏,“Orthogroup”、 “Drosophila_melanogaster” and “Homo_sapiens”。在表中找到 “FBgn0005648”,你会看到该基因属于orthogroup OG0001189,它在人类中有三个orthologues ENSG00000205022,ENSG00000100836,ENSG00000258643。
基因树
我们已经发现FBgn0005648在人类中有三个同源物。接下来我们要看一下基因树,看看我们是否同意这一点,并看看这三个正交物是如何产生的。用Dendroscope或基于网络的查看器打开Gene_Trees/OG0001189_tree.txt。

如果你习惯于推断和观察基因树,你会注意到OrthoFinder已经自动为你建立了树根–树根是在果蝇基因FBgn0005648上的。这使得它在快速检查基因树时非常方便,对于更复杂、更难解释的基因树尤其有用。
此外,默认的基因树没有支持值。毕竟,OrthoFinder已经将~121,000个基因分配到了正交组中,并在大约15分钟内为这些基因推断出了近18,000棵基因树! 我们将在后面的教程中讨论如何获得支持率值。
看一下基因树,我们可以看到发生了两个基因复制事件,一个是脊椎动物共有的,另一个是人类和小鼠共有的。这导致了一对三的直系同源关系,即所有三个人类基因与一个果蝇基因的关系同样密切。通常情况下,直系同源关系并不是一对一的,了解这一点很重要–你不希望花几个月的时间做 "直系同源 "的实验,但后来发现实际上有三个直系同源基因!
我们可以看一下FlyBase上这个基因的页面:http://flybase.org/reports/FBgn0005648.html。如果你下到 "直系亲属 "部分,然后看 “人类直系亲属”,你会发现唯一能确定所有这三个直系亲属的方法是基于树的方法Compara、 eggNOG、OrthoFinder和TreeFam。OrthoFinder是唯一一个你可以在自己的数据上运行的方法。基于分数的方法,如Hieranoid、Inparanoid、OMA和OrthoMCL只识别了一个,或没有识别这些正交物。基因树对于识别和解决这些复杂的关系特别重要。
基因复制事件
拥有基因树意味着OrthoFinder可以识别所有发生的基因复制事件。让我们来看看那些发生在脊椎动物共同祖先中的事件。OrthoFinder在Species_Tree/SpeciesTree_rooted_node_labels.txt文件中标注了物种树的节点,用Dendropscope打开这个文件,告诉它 “解释为节点标签”。我们将查看节点 “N1”,即脊椎动物的共同祖先(即D. rerio、T. rubripes、X. tropicalis、H. sapiens和M. musculus)。有两个文件提供了关于基因复制事件的细节。我们先在Dendroscope中打开Gene_Duplication_Events/SpeciesTree_Gene_Duplications_0.5_Support.txt。

这给出了一个基因复制事件的摘要。每个节点显示节点名称,后面是下划线,然后是物种树中映射到每个节点的支持良好的基因复制事件的数量。如果至少有50%的后代物种保留了复制基因的两个拷贝,则基因复制事件被认为是 "支持良好 "的。对于四足动物的共同祖先N1,有2458个这种支持良好的基因复制事件。我们可以在Gene_Duplication_Events/Duplications.tsv文件中看到这些基因复制事件的清单。下面是该文件中的几行,按其发生的物种树节点排序。

每个基因复制事件都与物种树节点、其发生的orthogroup/基因树以及该基因树中的节点相互参照。它还列出了由该基因复制事件产生的两个拷贝中的每一个的后代基因。我们可以为我们的FBgn0005648orthogroup检查这一点。为此,我们需要查看Resolved_Gene_Trees/OG0001189_tree.txt中的基因树。这个目录包含了标有节点的基因树。这些文件显示了OrthoFinder在推断直系亲缘关系和基因复制事件时对基因树的解释。它们可能与直接来自树推断步骤的原始基因树略有不同(可在Gene_Trees/)。为了得到已解决的基因树,OrthoFinder进行了复制-丢失-凝聚分析,以确定对树的更简明的解释。

从表上看,在节点n1有一个基因复制事件,两个拷贝在所有后代物种中都100%保留。看一下这棵树,第二个基因复制事件是在n10节点上,如果我们回到表格中,我们可以看到这个事件和Danio rerio的终端基因复制事件一样被列出。

如果你对基因复制事件感兴趣,那么这个表格就包含了大量的数据。在这六个物种中,OrthoFinder发现了34,065个基因复制事件,所有这些事件都与它们发生的物种树和基因树上的节点相互参照 在Duplications_per_Orthogroup.tsv和Duplications_per_Species_Tree_Node.tsv文件中,这些事件也按正交组和物种树节点进行了汇总,这两个文件都在Comparative_Genomics_Statistics/目录下。
Orthogroups
我们通常对群体性的物种比较感兴趣,也就是在一个物种支系之间而不是在一对物种之间进行比较。对多个物种的orthogly的概括就是orthogroup。就像orthologues 是一对物种的最后一个共同祖先中的一个基因的后代一样,orthogroup是一组物种中一个基因的后代的集合。OrthoFinder的每一棵基因树,例如上面的一棵,都是针对一个orthogroup的。如果我们希望orthogroup基因树包括所有成对的orthogroup,那么orthogroup基因树就是我们需要看的树。即使一个orthogroup内的一些基因可能是彼此的旁系物,如果我们试图把任何基因拿出来,那么我们也会把orthogroup拿出来。
因此,如果我们想对一组物种中的 "等价 "基因进行比较,我们需要对一个orthogroup中的基因进行比较。orthogroup在文件Orthogroups/Orthogroups.tsv中。这个表格每行有一个orthogroup,每列有一个spcies,从最大的orthogroup到最小的orthogroup排序。也有一个传统的OrthoMCL格式的文件。Orthogroups/Orthogroups.txt。
orthogroup sequence
对于每个orthogroup,在Orthogroup_Sequences/中都有一个FASTA文件,包含了该orthogroup的基因序列。
每一个OG包含的蛋白序列都记录在Orthogroups/Orthogroups.txt文件中,相应的每个OG fasta序列在Orthogroup_Sequences/OGxxxxxx.fa
(base) [yutao@myosin Results_Mar04]$ grep OG0011840 Orthogroups/Orthogroups.txt
OG0011840: PJADIDJD_01233 PJADIDJD_01234
(base) [yutao@myosin Results_Mar04]$ cat Orthogroup_Sequences/OG0011840.fa
>PJADIDJD_01233
MFGFFGGFRDFDRFKCGCGGWGGGCGGWGGRWNCDDWGGKWRNKWRCKRRDWC
>PJADIDJD_01234
MFGFFGGFRDFDRFKCGCGGWGGGCGGGWGGRWDFDCGGNWRNKWRCKRRDWC
Use the option, "-M msa -fg PREVIOUS_RESULTS_DIRECTORY". It will start from your already inferred orthogroups from the previous run but it will get OrthoFinder to select orthogroups that are single-copy in most species and then for each of these orthogroups use only the genes from those species that are single-copy to construct a concatenated MSA. This is what most studies do to infer a phylogeny, but OrthoFinder will do it automatically. It will use this to infer a species tree:
Species_Tree/SpeciesTree_rooted.txt
It will also produce two files to tell you the data used:
Species_Tree/Orthogroups_for_concatenated_alignment.txt
MultipleSequenceAlignments/SpeciesTreeAlignment.fa
You can get a fasta file for each orthogroup by running:
orthofinder -fg RESULTS_DIR -M msa -os
where RESULTS_DIR is the directory containing your orthogroups results files (e.g. Orthogroups.csv). I'll release an update soon so that you get these files from a default OrthoFinder run.
In RESULTS_DIR there is a file, SingleCopyOrthogroups.txt, which tells you which of these have all species present with exactly one gene per species. I think that answers your final question.
If you did want to find all the orthogroups with all species present (but no restriction on copy-number) then you can open the Orthogroups.GeneCount.csv file in excel and use that to identify them. E.g. for the example dataset I used the excel commands =COUNTIF(B3,">0"), =SUM(G2:J2) and =K2=4 as shown here: Orthogroups.GeneCount.xlsx
构建物种树方案
- 方案1:直接使用单拷贝OG串联构建进化树
- 方案2:单拷贝OG做的进化树,基因少,可以考虑90%以上的OG,构建物种树
- 方案3:选择在目标类群比例高,但是可能在外支不那么高的OG,确保目标系统发育比较准确的表示。
方案1:获得单拷贝OG的串联序列
- 选择单拷贝基因,分别进行多序列比对,然后串联比对结果构建系统发育树
(base) [yutao@myosin Results_Mar04]$ ls Single_Copy_Orthologue_Sequences
OG0000577.fa OG0000583.fa OG0000603.fa OG0000613.fa OG0000631.fa OG0000643.fa OG0000649.fa
OG0000580.fa OG0000584.fa OG0000609.fa OG0000614.fa OG0000639.fa OG0000644.fa
OG0000581.fa OG0000591.fa OG0000610.fa OG0000615.fa OG0000641.fa OG0000646.fa
OG0000582.fa OG0000593.fa OG0000611.fa OG0000625.fa OG0000642.fa OG0000647.fa
# 单个OG mafft比对和trimal
$ for i in *fa;do mafft --ep 0 --genafpair --maxiterate 1000 --thread 30 $i > ../single_copy_OG_msa/${i}_mafft.msa && trimal -in ../single_copy_OG_msa/${i}_mafft.msa -out ../single_copy_OG_msa/${i}_mafft_trimal.msa -gt 0.95 -cons 50 &>../single_copy_OG_msa/${i}_trimal.log ;done &
# 修改名称,便于串联
# Single_Copy_Orthologue_Sequences下的OG的序列顺序都是一样,即物种的顺序一样,因此可以选择一个序列的id作为标准,其他序列统一命名为这个,然后可以在AMAS中连到一起(没搞定)
# 直接从prokka蛋白序列找,137.id 顺序很重要
(base) [yutao@myosin single_copy_OG_msa]$ cat 137.id | while read id; do bioawk -c fastx -v id=$id '$name == id{n=split(FILENAME,a,"/");print id,a[n];exit}' /share/pasteur/yutao/Methanosarcinales_order_phylogene/Genome_integration/137MIMAG_High_quality_MAGs/137MAGs_faas/*faa;done|sed "s/_genomic.faa//g" > 137OG_ID_2_Genome.tsv &
# 这里第二列即是所有OG的新的序列名称
- 将prokka 蛋白ID的前面部分取出来作为名称,这样所有OG的都一样了
(base) [yutao@myosin 797OGs_atleastin_0.9MAGs_OG_sequences]$ for i in *trimal2;do bioawk -c fastx '{split($name,a,"_");print ">"a[1]"\n"$seq}' $i >${i}_rename.msa;done &
- 串联
(base) [yutao@myosin 797OGs_atleastin_0.9MAGs_OG_sequences]$ AMAS.py concat -i *rename.msa -f fasta -d aa -t 136MAGs_atleast0.9_OGs.msa -p 136MAGs_atleast0.9_OGs.msa.stat
- 将蛋白id修改成物种名称
gtdbtk) [yutao@myosin single_copy_OG_msa]$ cat rename_by_mapping_prtoid_with_taxid.sh
# (gtdbtk) [yutao@myosin single_copy_OG_msa]$ head 137OG_ID_2_Genome.tsv
#NADBEILJ_00247 GB_GCA_000306725.1
#CJPDHIMO_00953 GB_GCA_001602645.1
#CABJFEME_02449 GB_GCA_001714685.2
#DMFLKMLA_01936 GB_GCA_001896725.1
#(gtdbtk) [yutao@myosin single_copy_OG_msa]$ head OG0000609.fa_mafft_trimal.msa
#>NADBEILJ_01445
#GDSVIFVVKTTANQERSVANMITQVARKDHLDIRAIIAPDELKGYVLIEAPEPGVVEQAI
#QTVPHARTVVKGRSSIEEISHFLTPKPTVTGITEGAIIEITSGPFKGERARVKRVDEGHE
#EITVELFDAVVPIPITIRGDTVRVLRKEEE
#>CJPDHIMO_02580
#AETSIFVVKTTANQERSVANLIAQLARKEKYDITALLVPDVLKGYVLVEAKAPEIVEEAI
#QGVPHARTVIKGASSFGEVEHFLTPKPAVVGINEGAIVELISGPFKGEMARVKRVDIGKE
for msa in OG0000*_mafft_trimal.msa
do
line_num=1
cat 137OG_ID_2_Genome.tsv |while read i genome
do
bioawk -c fastx -v genome=$genome -v line_num=$line_num \
'FNR==line_num{print ">"genome"\n"$seq}' $msa
line_num=$(( $line_num + 1 ))
done >rename_trimal_msa/$msa
done
(base) [yutao@myosin single_copy_OG_msa]$ AMAS.py concat -i rename_trimal_msa/*msa -f fasta -d aa -t 137MAGs_25OGs.msa -p 137MAGs_25OGs.msa.stat
# 可以再修剪一次
(base) [yutao@myosin single_copy_OG_msa]$ time trimal -in 137MAGs_25OGs.msa -out 137MAGs_25OGs.msa_trimal.msa -gt 0.95 -cons 50 &>trimal.log
base) [yutao@myosin single_copy_OG_msa]$ ll 137MAGs_25OGs.msa_trimal.msa
-rw-r--r-- 1 yutao husn 683K Mar 8 19:45 137MAGs_25OGs.msa_trimal.msa
# 构树
(base) [yutao@myosin single_copy_OG_msa]$ nohup time raxmlHPC-PTHREADS -T 50 -x 888 -p 888 -f a -n boot -m PROTGAMMAAUTO -c 4 -e 0.001 -# 1000 -s 137MAGs_25OGs.msa_trimal.msa &>137MAGs_25OGs.msa_trimal.msa_raxml_bootstrap1000.log&
# 变成基因组名
(gtdbtk) [yutao@myosin 797OGs_atleastin_0.9MAGs_OG_sequences]$ Rename_newick_tip_label_by_mapping_table.r 136MAGs_atleast0.9_OGs_trimal.msa_fasttree_LG_model.newick /share/pasteur/yutao/Methanosarcinales_order_phylogene/Genome_integration/137MIMAG_High_quality_MAGs/136MAGs_6961OGs_ALE_results/Mapping_prot_vs_genome.tab 136MAGs_atleast0.9_OGs_trimal.msa_fasttree_LG_model_genome_name.newick
- 自带的命令
- Species tree: Using 329 orthogroups with minimum of 91.2% of species having single-copy genes in any orthogroup
(orthofinder) [yutao@myosin 137MAGs_orthofinder]$ nohup time orthofinder -M msa -fg Results_Mar04/ &>msa_fg_option.log&
方案2:在90%以上基因组的OG建树
- 1.可以通过"Orthogroups/Orthogroups.GeneCount.tsv"文件删选不同的OG构建系统发育树。例如方案2可以通过在excel中执行:
=IF(COUNTIF(B2:EG2,">0") >= 0.9*136,">=90%"),来筛选在90%以上基因组中的存在的OG。将OGID拷贝到一个list中 - 2.拷贝OG的fasta序列
(base) [yutao@myosin 136MAGs_atleastin_0.9MAGs_OGs]$ cat 797OGs_atleastin_0.9MAGs.ogid |while read i;do cp ../136MAGs_orthofinder/Results_Jun09/Orthogroup_Sequences/${i}.fa 797OGs_atleastin_0.9MAGs_OG_sequences;done
- 3.单个OG mafft比对和trimal
(base) [yutao@myosin 797OGs_atleastin_0.9MAGs_OG_sequences]$ for i in *fa;do mafft --ep 0 --genafpair --maxiterate 1000 --thread 50 $i > ${i}_mafft.msa && trimal -in ${i}_mafft.msa -out ${i}_mafft_trimal.msa -gt 0.95 -cons 50 ${i}_trimal.log ;done &
# 或者之前有跑过的直接拷贝即可
(base) [yutao@myosin 797OGs_atleastin_0.9MAGs_OG_sequences]$ cat ../797OGs_atleastin_0.9MAGs.ogid |while read i;do cp /share/pasteur/yutao/Methanosarcinales_order_phylogene/Genome_integration/137MIMAG_High_quality_MAGs/136MAGs_7073OGs_ALE_results/7073OGs_newicks/${i}.mft.trimal2 . ;done
- 4.更改序列名称,串联构树
将prokka 蛋白ID的前面部分取出来作为名称,这样所有OG的都一样了
(base) [yutao@myosin 797OGs_atleastin_0.9MAGs_OG_sequences]$ for i in *trimal2;do bioawk -c fastx '{split($name,a,"_");print ">"a[1]"\n"$seq}' $i >${i}_rename.msa;done &
# 串联多序列结果
(base) [yutao@myosin 797OGs_atleastin_0.9MAGs_OG_sequences]$ AMAS.py concat -i *rename.msa -f fasta -d aa -t 136MAGs_atleast0.9_OGs.msa -p 136MAGs_atleast0.9_OGs.msa.stat
# 再trimal一次
(base) [yutao@myosin 797OGs_atleastin_0.9MAGs_OG_sequences]$ time trimal -in 136MAGs_atleast0.9_OGs.msa -out 136MAGs_atleast0.9_OGs_trimal.msa -gt 0.95 -cons 50 &>trimal.log
# 构树
# iqtree
(base) [yutao@myosin 797OGs_atleastin_0.9MAGs_OG_sequences]$ nohup time iqtree -s 136MAGs_atleast0.9_OGs_trimal.msa -T 50 -B 1000 -m LG+G -wbtl &>136MAGs_atleast0.9_OGs_trimal.msa_iqtree.log &
# -T threads
# -B, --ufboot NUM Replicates for ultrafast bootstrap (>=1000)
# -m LG(SUBSTITUTION MODEL, protein)
# --wbtl Like --boot-trees but also writing branch lengths
# 或者fasttree
(base)$ conda activate gtdbtk
(gtdbtk) [yutao@myosin 797OGs_atleastin_0.9MAGs_OG_sequences]$ nohup time FastTree -lg -boot 1000 136MAGs_atleast0.9_OGs_trimal.msa 1> 136MAGs_atleast0.9_OGs_trimal.msa_fasttree_LG_model.newick 2>fastree.log&
常见问题
系统文件限制问题
(orthofinder) [yutao@myosin 137MIMAG_High_quality_MAGs]$ ulimit -n 20000
# 如果提示权限,退出再登录即可

简介

OrthoFinder: phylogenetic orthology inference for comparative genomics
Genome Biology, 2019
摘要
在此,我们提出了OrthoFinder方法的一个重大进展。它扩展了OrthoFinder的高精度直系同源簇推断,提供了直系同源簇的系统发育推断、有根基因树、基因复制事件、有根物种树和比较基因组学统计。每个输出结果都在适当的真实或模拟数据集上进行了基准测试,如果存在可比较的方法,OrthoFinder相当于或优于这些方法。此外,OrthoFinder是Quest for Orthologs基准测试中最准确的直系同源基因推断方法。最后,OrthoFinder的综合系统发育分析的速度和可扩展性与最快的、基于分数的启发式方法相当。OrthoFinder可在https://github.com/davidemms/OrthoFinder。
背景介绍
确定基因序列之间的系统发育关系是比较生物学研究的基础。它为理解地球上生命的进化和多样性提供了框架,并使生物体之间的生物知识得到推断。考虑到这一过程对生物研究的多个领域的核心重要性,已经开发了一系列不同的软件工具,试图在给定用户提供的基因序列集后确定这些关系[1,2,3]。这些软件工具大多试图通过启发式分析从全对全BLAST[4]搜索中获得的成对序列相似度分数(或期望值),或BLAST的加速替代品,如DIAMOND[5]或MMseqs2[6],推断基因序列之间的系统发育关系。广泛使用的方法包括InParanoid[7]、OrthoMCL[8]、OMA[9]和OrthoFinder[10],它们都采取了不同的方法来查询序列相似性分数,并且都产生了不同的输出结果–有些识别直系同源簇,有些识别直系和旁系,有些则两者都有。由于它们各自采用了不同的方法来分析序列相似性分数,所以每种方法在常用的基准数据库上表现出不同的性能特征[1, 11]。
对序列相似性分数的启发式分析历来被用来估计基因之间的系统发育关系,因为它在计算上是很容易的。使用这些方法的核心前提是,得分较高的序列对可能比得分较低的序列对在最近发生了分歧。因此,对成对序列相似性分数的启发式分析可用于估计基因组之间的系统发育关系[7,8,9, 12,13]。然而,这种基于分数的基因之间的系统发育关系的估计会受到多种因素的干扰。例如,基因之间可变的序列进化率经常导致假阳性和假阴性的错误[14,15](图1)。这种错误可以通过分析基因的系统发育树来缓解[17],因为系统发育树能够将可变的序列进化率(分支长度)与序列分化的顺序(树的拓扑结构)区分开来,从而阐明直系和旁系关系(图1)。

基于配对相似度评分的正体推断可能被可变的序列进化率所误导。 a 一个典型基因家族的系统发育树。b 物种A-基因2被误认为是物种B和C的基因的直系亲缘关系。 左侧:基因树的分支比例和真正的直系亲缘关系,可以从基因树上确定。右手边。基于与分支长度单调的基因相似性分数的互换最佳命中率(RBH),以及使用标准启发式方法从这些分数推断出的正交关系(使用RBH推断出的正交关系和从物种内命中率确定的共同正交关系比最接近的RBH更好[8,16])。FP,假阳性;FN,假阴性

OrthoFinder的工作流程。每个步骤使用的方法由箭头表示。已发表的算法以斜体字显示,后面有星号。与实心箭头相连的蓝色虚线表示额外的数据,用于进行实心箭头所示的转换。MSA,基于多序列排列的树推断;DLC,复制-丢失-凝聚。(a)使用最初的OrthoFinder算法进行正交组推断(正交组是所有被考虑的物种的最后一个共同祖先中的一个基因的后代集合)。(b) 基因树推断。© 物种树推断。(d) 物种树推断 (e) 基因树推断 (f) 对有根的基因树进行混合重叠+DLC分析,以推断正交物和基因重复事件。(g) 每个输入物种的基因的正交结果表的说明(四个主框)。其中的水平划分显示了每个物种对的正交物。(h) 基因复制事件表的说明,显示了映射到物种树上的基因复制事件的位置,在基因树中的位置,重复基因在采样物种中的保留百分比,以及基因复制事件的后代基因。(i) 比较基因组学统计

一组脊索动物物种的OrthoFinder分析摘要。Ciona intestinalis, Danio rerio, Oryzias latipes, Xenopus tropicalis, Gallus gallus, Monodelphis domestica, Mus musculus, Rattus norvegicus, Pan troglodytes, and Homo sapiens。条形图和热图包含每个物种的数据,与a中树中的相应物种对齐。 a 由STAG推断的物种树,由STRIDE扎根。e 含有每个物种对的正交组数量的热图(右上)和每个物种之间的正交组(左下)。 f 两个物种,即C. intestinalis和H. sapiens,相对于所有其他物种的正交组倍数。g 物种树每个末端分支上的基因复制事件的数量。h 物种树每个分支上的复制和保留在所有后代物种的数量。OG,正组;sp.,物种;spp.,物种(复数);dups.,基因复制事件
已经开发了许多基于树的直系ton在线数据库,包括PhylomeDB[18]、Ensembl-Compara[19]、EggNOG[20]和TreeFam[21]。这些使用率很高的资源为用户提供了使用系统发育树探索基因进化历史的能力,比单纯的成对正交关系和旁系关系提供了更完整的图像。使用标准的基准方法对这些方法进行比较分析,发现这些在线数据库和基于分数的软件工具的正交检测准确率没有明显差异[1],表明系统发育方法的优势还没有完全实现。此外,这些在线数据库背后的管道和方法一般不提供给用户来运行他们自己的分析。因此,需要一个自动化的软件工具,有效地利用系统发育的方法来提高准确性,但又具有基于分数的启发式方法的易用性、速度和可扩展性。
虽然从基因序列中进行系统发育正交推断的自动软件工具是一个重要的目标,但这种方法的实施却有几个技术上的挑战。这些挑战包括以下内容。(1)在与基于分数的启发式方法竞争的时间尺度内,推断出一套完整的物种的所有基因树;(2)自动将这些基因树生根,使其能够被正确解释[22],而不需要用户事先知道生根的物种树;(3)解释基因树以识别基因复制事件、正交物和旁系物,同时对基因复制、丢失、不完整的系谱排序和基因树不准确等过程保持稳健。如果这些挑战能够以资源和时间效率的方式得到解决,那么这样的系统发育方法将为正交推断提供一个步骤的改变,使之从基于相似度分数的系统发育关系估计过渡到基因间的系统发育关系的划分。
上述的一些挑战已经被一系列的生物信息学方法孤立地解决了。例如,有一系列从用户提供的基因序列中识别基因正交组的方法[8,9,10,12,23],以及各种可以从这些正交组推断出树的基因树推断方法[24,25,26,27,28]。同样,有一系列从基因树推断直系亲属的方法,这些方法在可扩展性和准确性方面也有不同[29,30,31,32]。然而,其他的关键挑战没有现有的解决方案。例如,从一组物种蛋白质组推断出一套完整的有根基因树将是一个复杂的、多步骤的过程,一般需要事先了解物种树。同样,从基因树推断正交物的方法也不存在,这些方法对不完整的系谱排序和基因树推断错误等过程具有鲁棒性,同时也可以扩展到数百个物种的全基因组正交推断所需的大规模分析。因此,需要解决大量的技术挑战,以实现对基因之间系统发育关系的全自动、准确和高效的系统发育划定。
在此,我们对OrthoFinder进行了重大更新,解决了这些挑战,并大大扩展了原有方法的范围。更新后的OrthoFinder与原来的实现方法[10]一样,确定了正交组,但随后利用这些正交组推断出所有正交组的基因树,并对这些基因树进行分析以确定有根的物种树。随后,该方法在完整的基因树集中识别所有的基因复制事件,并在物种树的背景下分析这些信息,以提供基因树和物种树层面的基因复制事件分析。最后,该方法对所有这些系统发育信息进行分析,以确定所有物种之间完整的正交物集,并提供广泛的比较基因组学统计。完整的OrthoFinder系统发育正交推断方法是准确的、快速的、可扩展的和可定制的,只需一个命令就可以完成,只需使用蛋白质序列作为输入。
OG功能注释
每个OG通常有多个物种的多个蛋白,有两种方法来对OG进行功能注释:第一种简单的方法是选择OG中最长的作为代表,作为最终的注释结果;另外一种是对OG中每一个都进行注释,然后选择最多的作为注释结果。这里对每个蛋白的注释可以使用eggnog的结果。
1.选择每个OG最长的蛋白作为注释
- 蛋白长度文件按
(base) [yutao@myosin Results_Mar04]$ bioawk -c fastx '{print $name"\t"length($seq)}' ../../137MAGs_faas/*faa >137MAGs_protein_faa_length.tsv &
- eggnog注释文件
(base) [yutao@myosin Results_Mar04]$ cat ../../137MAGs_eggnogs/annotations/*emapper.annotations >137MAGs_protein_eggnog_annotations.tsv
- Eggnog_Function_annotation_for_each_OrthoGroup_by_Max_Length_Protein.py
# select protein of max length OG member function anotation of each OrthoGroup
import sys,os
from collections import Counter
def helpDoc():
print('Usage: python3 $0 <Orthogroups.txt> <Each protein length> <Merged eggnog table> <out>')
print('\tOrthogroups.txt:')
print('\tOG0011839: AJKKFJJO_01576 AJKKFJJO_01819')
print('\tOG0011840: PJADIDJD_01233')
print('\tEach protein length: protein_id\tlength')
print('\tMerged eggnog table: Merged eggnog annotation table')
if len(sys.argv) != 5:
helpDoc()
quit('error input!')
f_og = sys.argv[1]
f_pro_len = sys.argv[2]
f_ant = sys.argv[3]
f_out = sys.argv[4]
# 读入蛋白id和长度表格,存成字典
Prot_len_dict = {}
with open(f_pro_len,'r') as f:
for line in f:
line = line.strip()
protid, protlen = line.split('\t')
if protid not in Prot_len_dict:
Prot_len_dict[protid] = protlen
#print(Annt_dict)
# 2.读入OG的文件,包含每个OG id及其所属蛋白的信息
## 将OG id作为key,后面的蛋白id作为值存成字典
OG_dict = {}
with open(f_og, 'r') as f:
for line in f:
line = line.strip()
(ogid, prots) = (line.split(":")[0], line.split(":")[1].split())
OG_dict[ogid] = prots
#print(OG_dict)
# 获得每一个OG对应蛋白的长度,取出最长的存起来
# 遍历每一个OG,取出每一个OG的成员,取出最大长度的作为OG的代表
OG_rep_prot = {} # 存储每一个OG代表性的蛋白id
for ogid in OG_dict:
prots = OG_dict[ogid] # 取出OG每个proid
prot_num = len(prots)
if prot_num == 1: # 如果只要一个则直接指定
OG_rep_prot[ogid] = ''.join(prots) # 列表变成字符串
else:
# 遍历每个protid拿到对应的注释信息
prot_lens = {} # 将同一个OG下所有蛋白的长度存成字典
for protid in prots:
if protid in Prot_len_dict:
prot_len = Prot_len_dict[protid]
prot_lens[protid] = prot_len
else:
print('%s not in protein length table!')
sorted_x = sorted(prot_lens.items(), key=lambda x: x[1], reverse=True)
rep_prot = sorted_x[0][0] # 第一个是最长的
OG_rep_prot[ogid] = rep_prot
#print(OG_rep_prot)
# 将所有蛋白的eggnog注释存成字典
Annt_dict = {}
with open(f_ant, 'r') as f:
for line in f: # 正式对每一条query的注释结果进行解析
line = line.strip() # 去掉两端空白符号
if not line.startswith('#'):
(query,seed_ortholog,evalue,score,eggNOG_OGs,max_annot_lvl,
COG_category,Description,Preferred_name,GOs,EC,KEGG_ko,KEGG_Pathway,
KEGG_Module,KEGG_Reaction,KEGG_rclass,BRITE,KEGG_TC,CAZy,BiGG_Reaction,PFAMs) = line.split('\t') # 制表符分隔成不同列
if query not in Annt_dict:
Annt_dict[query] = line
# 给每一个OG最长的蛋白找到注释
if os.path.exists(f_out):
os.remove(f_out)
with open(f_out, 'a') as f:
header = 'OG\trep_query\tseed_ortholog\tevalue\tscore\teggNOG_OGs\tmax_annot_lvl\tCOG_category\tDescription\tPreferred_name\tGOs\tEC\tKEGG_ko\tKEGG_Pathway\tKEGG_Module\tKEGG_Reaction\tKEGG_rclass\tBRITE\tKEGG_TC\tCAZy\tBiGG_Reaction\tPFAMs\n'
f.write(header)
for ogid in OG_rep_prot:
protid = OG_rep_prot[ogid]
if protid in Annt_dict:
annt = Annt_dict[protid]
else:
annt = 'eggnog_NA'
# 格式化输出
line = ('%s\t%s\n' % (ogid, annt))
f.write(line)

开放原子开发者工作坊旨在鼓励更多人参与开源活动,与志同道合的开发者们相互交流开发经验、分享开发心得、获取前沿技术趋势。工作坊有多种形式的开发者活动,如meetup、训练营等,主打技术交流,干货满满,真诚地邀请各位开发者共同参与!
更多推荐
 4
4 0
0- 0



 已为社区贡献5条内容
已为社区贡献5条内容

所有评论(0)