生物分子体系结构预测开源模型RoseTTAFold All-Atom的conda环境部署及使用
本文提供了生物体系结构预测开源模型RoseTTAFold All-Atom的Conda安装及使用体验。
| 欢迎浏览我的CSND博客! Blockbuater_drug …点击进入 |
|---|
文章目录
前言
本文提供了生物分子体系结构预测开源模型RoseTTAFold All-Atom的Conda安装及使用体验。
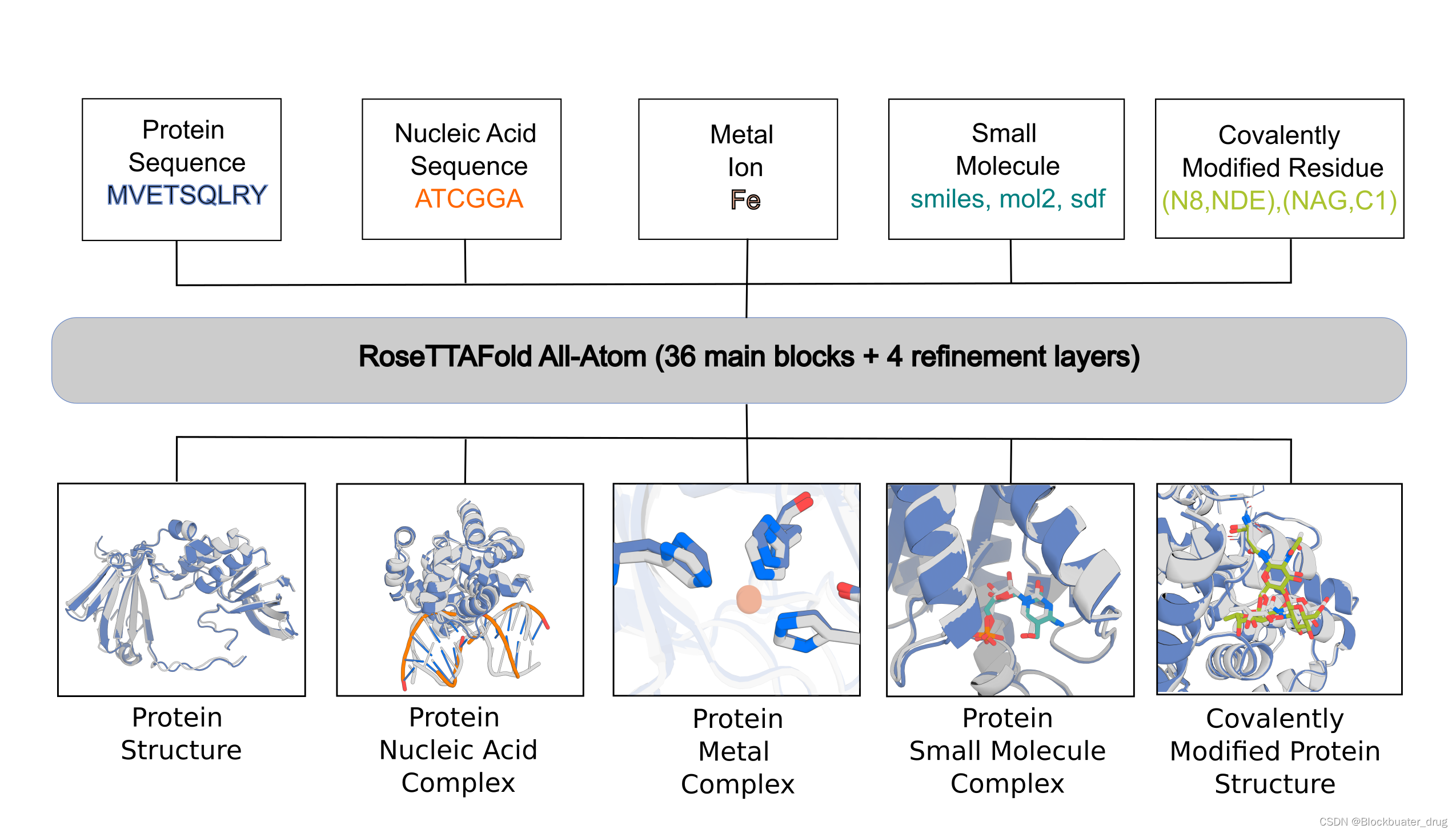
一、RoseTTAFold All-Atom(RFAA)是什么?
github代码:https://github.com/baker-laboratory/RoseTTAFold-All-Atom
science介绍文章:https://www.science.org/doi/10.1126/science.adl2528
science文章的预印本:https://www.biorxiv.org/content/10.1101/2023.10.09.561603v1.full.pdf
2024年3月7日,Science期刊报道了来自David Baker团队的蛋白质设计工具更新:RoseTTAFold All-Atom(RFAA),标题为Generalized biomolecular modeling and design with RoseTTAFold All-Atom。RFAA可以将氨基酸和 DNA 碱基的基于残基的表示与所有其他基团的原子表示相结合,从而对包含蛋白质、核酸、小分子、金属和给定序列和化学结构的共价修饰的组件进行建模。
似乎看到了AlphaFold-latest的影子。。。。
RFAA通过对扩散去噪任务进行微调,它通过直接在小分子和其他非蛋白质分子周围构建蛋白质结构,来生成结合口袋。将结构预测推广到所有生物分子。Baker 表示:「原则上,经过更多样化数据集训练的网络应该能够更好地进行泛化。」他补充说,研究人员计划让该网络取代任务特定版本的 RoseTTAFold。
具体来说,研究人员根据 RoseTTAFold2(RF2)蛋白质结构预测网络对网络架构进行建模,该网络可以接受 1D 序列信息、来自同源模板的 2D 成对距离信息和 3D 坐标信息,并通过许多隐藏层迭代改进预测结构。与蛋白质和核酸序列不同,分子图是排列不变的,因此,无论小分子元素标记顺序如何,网络都应该做出相同的预测。AF2 和 RF2 中,氨基酸和碱基的序列顺序是通过相对位置编码来表示的;对于原子,该团队省略了这样的编码并利用网络注意力机制的排列不变性。
研究人员还修改了坐标更新:在 AF2 和 RF2 中,蛋白质残基由 C 坐标和 N-C -C 刚性框架 α 的方向 α 表示,并且沿着 3D 轨迹,网络生成每个框架方向的旋转更新,以及每个坐标的平移更新。为了在 RFAA 中概括这一点,重原子坐标被添加到 3D 轨迹中,并仅根据对其位置的预测平移更新独立移动。因此,在输入后,整个系统立即被表示为氨基酸残基、核酸碱基以及自由移动原子的气体,它通过网络的许多块连续转化为物理上合理的组装结构。对于指导参数优化的损失函数,研究人员开发了 AF2 中引入的帧对齐点误差(FAPE)损失的全原子版本,通过根据其键合邻居的身份定义任意分子中每个原子的坐标系,与基于残基的 FAPE 一样,连续对齐每个坐标系并计算周围原子的坐标误差。除了原子坐标之外,网络还可以预测原子和残基置信度 (pLDDT) 和成对置信度 (PAE) 指标,从而能够识别高质量的预测。
简言之,RFAA可以预测所有主要的生物分子体系,包括与大分子相互作用的其他分子,可以用来做药物分子设计与生成。

 看看RFAA的效果如何:
看看RFAA的效果如何:


二、安装步骤
安装环境:Ubuntu 22.04, CUDA runtime版本11.8,GCC 11.4。
1. 安装mamba(非必须的,conda也可以)
wget "https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-$(uname)-$(uname -m).sh"
bash Mambaforge-$(uname)-$(uname -m).sh
2. 下载RoseTTAFold-All-Atom
重新打开一个Terminal:
git clone https://github.com/baker-laboratory/RoseTTAFold-All-Atom
cd RoseTTAFold-All-Atom
3. 创建conda环境并安装
mamba env create -f environment.yaml
问题:可能会卡在 Installing pip dependencies
解决:退出。进入环境,然后pip install -r list.txt安装相应内容即可。
conda activate RFAA
4. 安装SE3T
cd rf2aa/SE3Transformer
pip install --no-cache-dir -r requirements.txt
python3 setup.py install
5. 准备cs-blast
bash install_dependencies.sh
6. 安装signalp6
下载并安装,在conda RFAA_env环境中运行:
signalp6-register signalp-6.0h.fast.tar.gz
修改模型参数文件的名称,执行如下:
mv $CONDA_PREFIX/lib/python3.10/site-packages/signalp/model_weights/distilled_model_signalp6.pt $CONDA_PREFIX/lib/python3.10/site-packages/signalp/model_weights/ensemble_model_signalp6.pt
输入signalp6,查看安装,输出:

7. 下载序列和模板数据库
下载数据库,放置在RoseTTAFold-All-Atom文件夹;如果置于其他文件夹,需要与base.yaml 和make_msa.sh的参数路径匹配。
遇到的问题1: 数据库下载慢
解决方法: conda安装的情况下,数据库可以使用软连接到其他来源,可以连接到AF2或者RF2的数据库。
uniref30 [46GB]
wget http://wwwuser.gwdg.de/~compbiol/uniclust/2020_06/UniRef30_2020_06_hhsuite.tar.gz
mkdir -p UniRef30_2020_06
tar xfz UniRef30_2020_06_hhsuite.tar.gz -C ./UniRef30_2020_06
BFD [272GB]
wget https://bfd.mmseqs.com/bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt.tar.gz
mkdir -p bfd
tar xfz bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt.tar.gz -C ./bfd
structure templates [87GB]
包含pdb100_2021Mar03_a3m.ffdata, pdb100_2021Mar03_a3m.ffindex, pdb100_2021Mar03_cs219.ffdata, pdb100_2021Mar03_cs219.ffindex, pdb100_2021Mar03_hhm.ffdata, pdb100_2021Mar03_hhm.ffindex, pdb100_2021Mar03_pdb.ffdata, pdb100_2021Mar03_pdb.ffindex 一共8个文件,解压后~300 GB。
wget https://files.ipd.uw.edu/pub/RoseTTAFold/pdb100_2021Mar03.tar.gz
tar xfz pdb100_2021Mar03.tar.gz
8. 运行RFAA
运行方法:
python -m rf2aa.run_inference --config-name {your inference config}
其中,your inference config的示例文件位于 rf2aa/config/inference目录,当前有5种config模板文件供参考:
protein:单体monomer
protein_complex_sm: 蛋白-小分子复合物
nucleic_acid:核酸
protein_na_sm:蛋白-核酸-小分子复合物
covalent:蛋白-共价小分子复合物
相应的fasta文件位于examples文件。
使用时修改或者建立相应的config文件,然后将fasta文件置于examples文件夹相应的位置。
8.0 运行可能遇到的问题及解决方法
遇到的问题1: 报错 sequence ss_pred contains no residues
 解决方法: 类似于 RosettaFold的解决方法
解决方法: 类似于 RosettaFold的解决方法
wget https://ftp.ncbi.nlm.nih.gov/blast/executables/legacy.NOTSUPPORTED/2.2.26/blast-2.2.26-x64-linux.tar.gz
tar -zxvf blast-2.2.26-x64-linux.tar.gz
修改make_ss.sh文件:在开始位置添加export BLASTMAT=绝对路径
#!/bin/bash
# From: https://github.com/RosettaCommons/RoseTTAFold
export BLASTMAT=$FULLPATHTO/blast-2.2.26/data/
遇到的问题2: 12GB,提示显存不足,退出
解决方法: 将pytorch的 max_split_size_mb设置小一些,加进环境变量
export PYTORCH_CUDA_ALLOC_CONF=max_split_size_mb: 1024
5种运行实例如下:
8.1 运行单体蛋白(monomer)结构预测
python -m rf2aa.run_inference --config-name protein
protein.yaml文件内容如下:
defaults:
- base
job_name: "7u7w_protein"
protein_inputs:
A:
fasta_file: examples/protein/7u7w_A.fasta
435AA,运行结果如下;RMSD=1.432,

8.2 运行蛋白-小分子复合物结构预测
python -m rf2aa.run_inference --config-name protein_complex_sm
结果展示如下,很好的找到了小分子的结合位置。

8.3 运行蛋白-核酸复合物结构预测
python -m rf2aa.run_inference --config-name nucleic_acid
运行结果如下,RMSD=1.688

8.4 运行蛋白-核酸-小分子多聚体结构预测
python -m rf2aa.run_inference --config-name protein_na_sm
结果展示:

8.5 运行蛋白-共价结合小分子结构预测
python -m rf2aa.run_inference --config-name covalent
运行30min,结果:

总结
本文提供了生物体系结构预测开源模型RoseTTAFold All-Atom的Conda安装及使用体验。
参考资料
- https://github.com/baker-laboratory/RoseTTAFold-All-Atom
- https://www.science.org/doi/10.1126/science.adl2528
- https://zhuanlan.zhihu.com/p/685998369
| 欢迎浏览我的CSND博客! Blockbuater_drug …点击进入 |
|---|

开放原子开发者工作坊旨在鼓励更多人参与开源活动,与志同道合的开发者们相互交流开发经验、分享开发心得、获取前沿技术趋势。工作坊有多种形式的开发者活动,如meetup、训练营等,主打技术交流,干货满满,真诚地邀请各位开发者共同参与!
更多推荐
 58
58 0
0- 0



 已为社区贡献14条内容
已为社区贡献14条内容

所有评论(0)