
基于功能预测数据下游分析的R包ggpicrust2
写在前面总体来讲,作者这个R包提供了基因和功能的对应表格和函数,方便不了解功能的朋友进行尝试。其次提供了几种展示功能差异的可视化办法,作者尝试用组合图展示,尝试和大家已经有的东西区分开,这是一种有益的尝试。但是整理可视化效果和布局都有待进一步提升。但是值得大家进行尝试学习。第一作者:Chen Yang仓库地址:https://github.com/cafferychen777/ggpicrust2
写在前面
总体来讲,作者这个R包提供了基因和功能的对应表格和函数,方便不了解功能的朋友进行尝试。其次提供了几种展示功能差异的可视化办法,作者尝试用组合图展示,尝试和大家已经有的东西区分开,这是一种有益的尝试。但是整理可视化效果和布局都有待进一步提升。但是值得大家进行尝试学习。
第一作者:
Chen Yang
仓库地址:
https://github.com/cafferychen777/ggpicrust2#citation
文章地址:
https://arxiv.org/abs/2303.10388
本文代码数据地址:https://github.com/taowenmicro/Rcoding/tree/main/ggpicrust2_%E5%8A%9F%E8%83%BD%E5%88%86%E6%9E%90
作者摘要
微生物组研究现在已经超越了对样本中微生物的组成分析。来自大规模人体微生物组研究的越来越多的证据表明,肠道微生物组变化的功能后果可能为研究其对炎症和免疫反应的影响提供更大的力量。虽然16S rRNA分析是最流行的一种成本效益高的方法,用于描述微生物组成,但标记基因测序无法直接提供社区成员基因组中存在的功能基因的信息。已经开发了生物信息学工具,利用16S rRNA基因数据来预测微生物组功能。其中,PICRUSt2已成为最流行的功能概况预测工具之一,可生成整个群落通路丰度。然而,目前没有最先进的推断工具可用于测试比较组之间通路丰度的差异。我们开发了ggpicrust2,这是一个R软件包,可进行广泛的差异丰度(DA)分析,并提供可出版的可视化。

安装R包
安装R包出现了lefser包的错误,所以这里我先安装这个依赖,然后再安装目标R包;
#--安装R包
BiocManager::install("lefser")
devtools::install_github('cafferychen777/ggpicrust2')使用三人成师数据演示
数据内容全部放到github中,大家可以下载重复.下面这个代码是一个流程性的代码,一条解决全部问题,但是往往不容易弄清楚作者是怎么做的。所以这个运行结束后面还有逐条运行。
#--实例
library(readr)
library(ggpicrust2)
library(tibble)
library(tidyverse)
library(ggprism)
library(patchwork)
map <-
read_csv(
"./map_16s.csv"
)
map$...1 = NULL
group <- "Group"
library(R.utils)
gunzip("./picrust2_out_pipeline/EC_metagenome_out/pred_metagenome_unstrat.tsv.gz", remove = TRUE)
daa_results_list <-
ggpicrust2(
file = "./picrust2_out_pipeline/EC_metagenome_out/pred_metagenome_unstrat.tsv",
metadata = map,
group = "Group",
pathway = "KO",
daa_method = "LinDA",
p_values_bar = TRUE,
p.adjust = "BH",
ko_to_kegg = TRUE,
order = "pathway_class",
select = NULL,
reference = NULL
)逐步运行差异分析—出图
这里使用了ALDEx2方法,还有一些其他方法,这里作者主要参考的NC上面14中差异方法, 具体大家可以自己查看。
#If you want to analysis kegg pathway abundance instead of ko within the pathway. You should turn ko_to_kegg to TRUE.
#The kegg pathway typically have the more explainable description.
#metadata should be tibble.
library(readr)
library(ggpicrust2)
library(tibble)
library(tidyverse)
library(ggprism)
library(patchwork)
metadata <-
map
kegg_abundance <-
ko2kegg_abundance(
"./picrust2_out_pipeline/KO_metagenome_out/pred_metagenome_unstrat.tsv"
)
group <- "Group"
#--进行差异分析
daa_results_df <-
pathway_daa(
abundance = kegg_abundance,
metadata = map,
group = group,
daa_method = "ALDEx2",
select = NULL,
reference = NULL
)柱状图组合图可视化
这里注意,通路显著的数量较多的话,可视化不出来,所以这个工具现在不是很好用,后面作者应该会进行很多更新,但是目前有基于功能预测的进一步分析工具,作者已经在努力了,加油。
daa_sub_method_results_df <-
daa_results_df[daa_results_df$method == "ALDEx2_Kruskal-Wallace test", ]
daa_annotated_sub_method_results_df <-
pathway_annotation(pathway = "KO",
daa_results_df = daa_sub_method_results_df,
ko_to_kegg = TRUE)
Group <-
metadata$Group # column which you are interested in metadata
# select parameter format in pathway_error() is c("ko00562", "ko00440", "ko04111", "ko05412", "ko00310", "ko04146", "ko00600", "ko04142", "ko00604", "ko04260", "ko04110", "ko04976", "ko05222", "ko05416", "ko00380", "ko05322", "ko00625", "ko00624", "ko00626", "ko00621")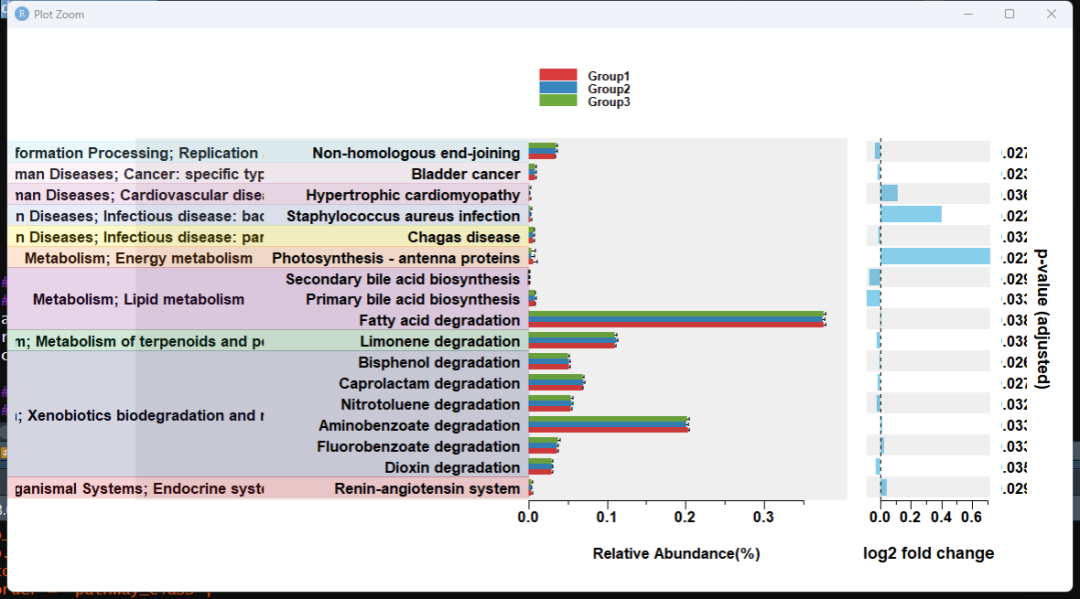
热图和排序图展示
有这两个图形的演示,可以绘制。
# 展示组合柱状图和误差图#--------
daa_results_list <-
pathway_errorbar(
abundance = kegg_abundance,
daa_results_df = daa_annotated_sub_method_results_df,
Group = Group,
p_values_threshold = 0.04,
order = "pathway_class",
select = NULL,
ko_to_kegg = TRUE,
p_value_bar = TRUE,
colors = NULL,
x_lab = "pathway_name"
)
# 展示热图#---------
# Create example functional pathway abundance data
abundance_example <- matrix(rnorm(30), nrow = 10, ncol = 3)
rownames(abundance_example) <- paste0("Sample", 1:10)
colnames(abundance_example) <- c("PathwayA", "PathwayB", "PathwayC")
# Create example metadata
# Please change your sample id's column name to sample_name
metadata_example <- data.frame(sample_name = rownames(abundance_example),
group = factor(rep(c("Control", "Treatment"), each = 5)))
heatmap_plot <- ggpicrust2::pathway_heatmap(t(abundance_example), metadata_example, "group")#--展示排序图#---------
# Create example functional pathway abundance data
abundance_example <- data.frame(A = rnorm(10), B = rnorm(10), C = rnorm(10))
# Create example metadata
metadata_example <- tibble::tibble(sample_id = 1:10,
group = factor(rep(c("Control", "Treatment"), each = 5)))
pca_plot <- ggpicrust2::pathway_pca(t(abundance_example), metadata_example, "group")
猜你喜欢iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读
瓜分20万奖金 获得内推名额 丰厚实物奖励 易参与易上手
更多推荐

 1
1 0
0- 0



 已为社区贡献8条内容
已为社区贡献8条内容

所有评论(0)